

YbxF and YlxQ are bacterial homologs of L7Ae and bind K-turns but not K-loops
- PMID: 22355167
- PMCID: PMC3312563
- DOI: 10.1261/rna.031518.111
YbxF and YlxQ are bacterial homologs of L7Ae and bind K-turns but not K-loops
Abstract
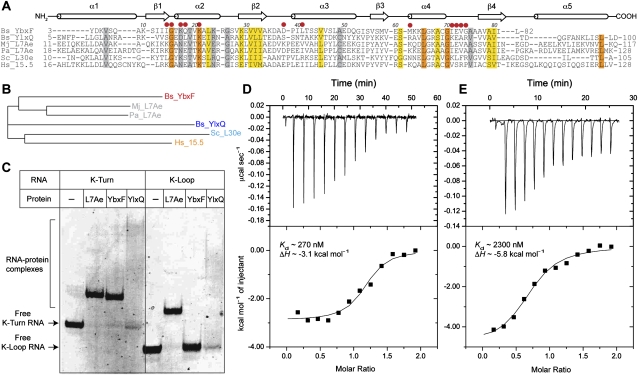
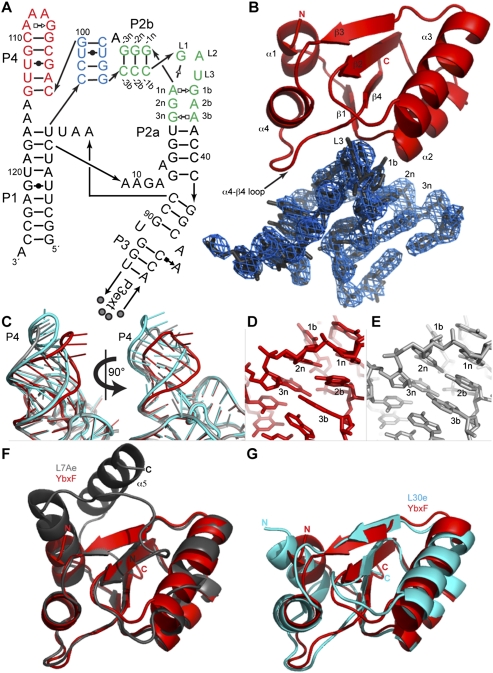
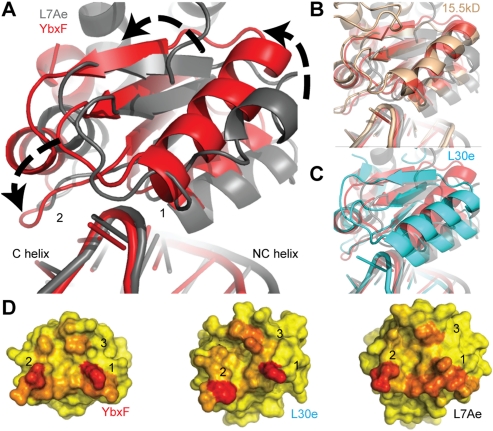
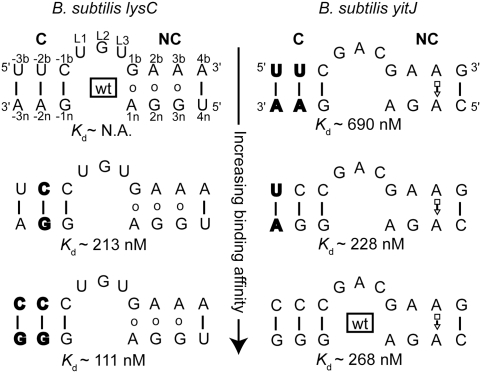
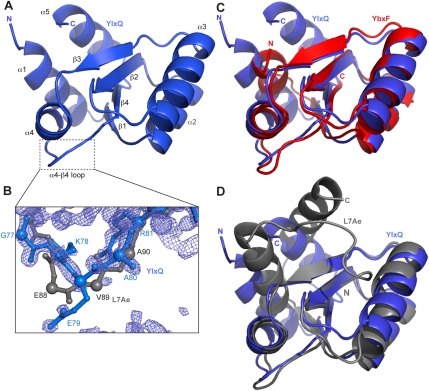
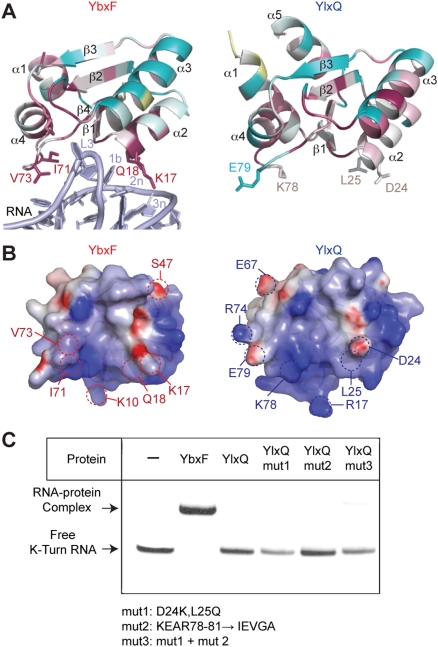
The archaeal protein L7Ae and eukaryotic homologs such as L30e and 15.5kD comprise the best characterized family of K-turn-binding proteins. K-turns are an RNA motif comprised of a bulge flanked by canonical and noncanonical helices. They are widespread in cellular RNAs, including bacterial gene-regulatory RNAs such as the c-di-GMP-II, lysine, and SAM-I riboswitches, and the T-box. The existence in bacteria of K-turn-binding proteins of the L7Ae family has not been proven, although two hypothetical proteins, YbxF and YlxQ, have been proposed to be L7Ae homologs based on sequence conservation. Using purified, recombinant proteins, we show that Bacillus subtilis YbxF and YlxQ bind K-turns (K(d) ~270 nM and ~2300 nM, respectively). Crystallographic structure determination demonstrates that both YbxF and YlxQ adopt the same overall fold as L7Ae. Unlike the latter, neither bacterial protein recognizes K-loops, a structural motif that lacks the canonical helix of the K-turn. This property is shared between the bacterial and eukaryal family members. Comparison of our structure of YbxF in complex with the K-turn of the SAM-I riboswitch and previously determined structures of archaeal and eukaryal homologs bound to RNA indicates that L7Ae approaches the K-turn at a unique angle, which results in a considerably larger RNA-protein interface dominated by interactions with the noncanonical helix of the K-turn. Thus, the inability of the bacterial and eukaryal L7Ae homologs to bind K-loops probably results from their reliance on interactions with the canonical helix. The biological functions of YbxF and YlxQ remain to be determined.
Figures
Similar articles
-
Signature amino acids enable the archaeal L7Ae box C/D RNP core protein to recognize and bind the K-loop RNA motif.RNA. 2010 Jan;16(1):79-90. doi: 10.1261/rna.1692310. Epub 2009 Nov 19. RNA. 2010. PMID: 19926724 Free PMC article.
-
The archaeal sRNA binding protein L7Ae has a 3D structure very similar to that of its eukaryal counterpart while having a broader RNA-binding specificity.J Mol Biol. 2004 Sep 17;342(3):757-73. doi: 10.1016/j.jmb.2004.07.046. J Mol Biol. 2004. PMID: 15342235
-
Comparative analysis of the 15.5kD box C/D snoRNP core protein in the primitive eukaryote Giardia lamblia reveals unique structural and functional features.Biochemistry. 2011 Apr 12;50(14):2907-18. doi: 10.1021/bi1020474. Epub 2011 Mar 18. Biochemistry. 2011. PMID: 21366326
-
The kink-turn in the structural biology of RNA.Q Rev Biophys. 2018 Jan;51:e5. doi: 10.1017/S0033583518000033. Q Rev Biophys. 2018. PMID: 30912490 Review.
-
The Kink Turn, a Key Architectural Element in RNA Structure.J Mol Biol. 2016 Feb 27;428(5 Pt A):790-801. doi: 10.1016/j.jmb.2015.09.026. Epub 2015 Oct 29. J Mol Biol. 2016. PMID: 26522935 Free PMC article. Review.
Cited by
-
Single-molecule observation of the induction of k-turn RNA structure on binding L7Ae protein.Biophys J. 2012 Dec 19;103(12):2541-8. doi: 10.1016/j.bpj.2012.11.006. Epub 2012 Dec 18. Biophys J. 2012. PMID: 23260056 Free PMC article.
-
Unboxing the T-box riboswitches-A glimpse into multivalent and multimodal RNA-RNA interactions.Wiley Interdiscip Rev RNA. 2020 Nov;11(6):e1600. doi: 10.1002/wrna.1600. Epub 2020 Jul 6. Wiley Interdiscip Rev RNA. 2020. PMID: 32633085 Free PMC article. Review.
-
Structure and folding of a rare, natural kink turn in RNA with an A*A pair at the 2b*2n position.RNA. 2012 Jun;18(6):1257-66. doi: 10.1261/rna.032409.112. Epub 2012 Apr 26. RNA. 2012. PMID: 22539525 Free PMC article.
-
The plasticity of a structural motif in RNA: structural polymorphism of a kink turn as a function of its environment.RNA. 2013 Mar;19(3):357-64. doi: 10.1261/rna.036657.112. Epub 2013 Jan 16. RNA. 2013. PMID: 23325110 Free PMC article.
-
Structure and mechanism of the T-box riboswitches.Wiley Interdiscip Rev RNA. 2015 Jul-Aug;6(4):419-33. doi: 10.1002/wrna.1285. Epub 2015 May 8. Wiley Interdiscip Rev RNA. 2015. PMID: 25959893 Free PMC article. Review.
References
-
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289: 905–920 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases