

Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling
- PMID: 22286106
- PMCID: PMC3454471
- DOI: 10.1016/j.cellsig.2012.01.008
Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling
Abstract
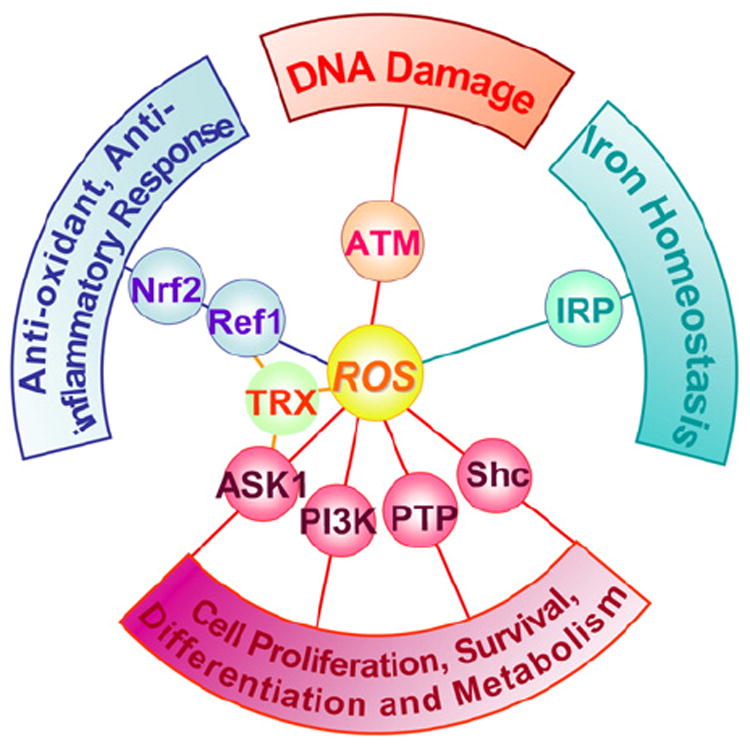
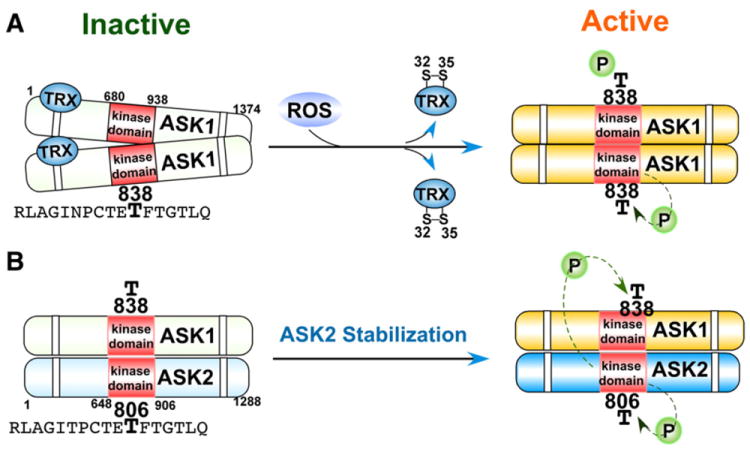
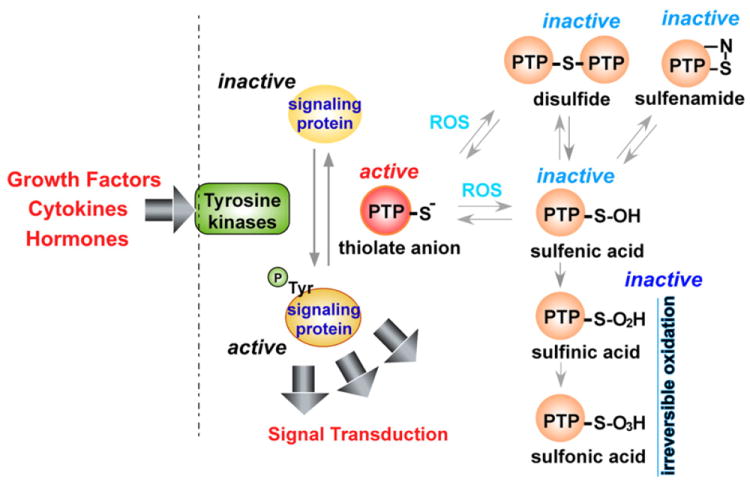
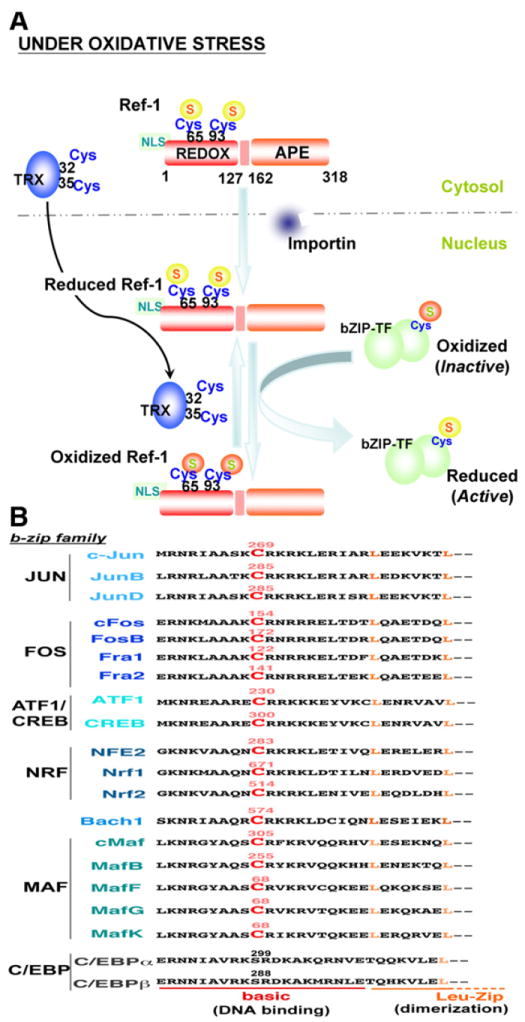
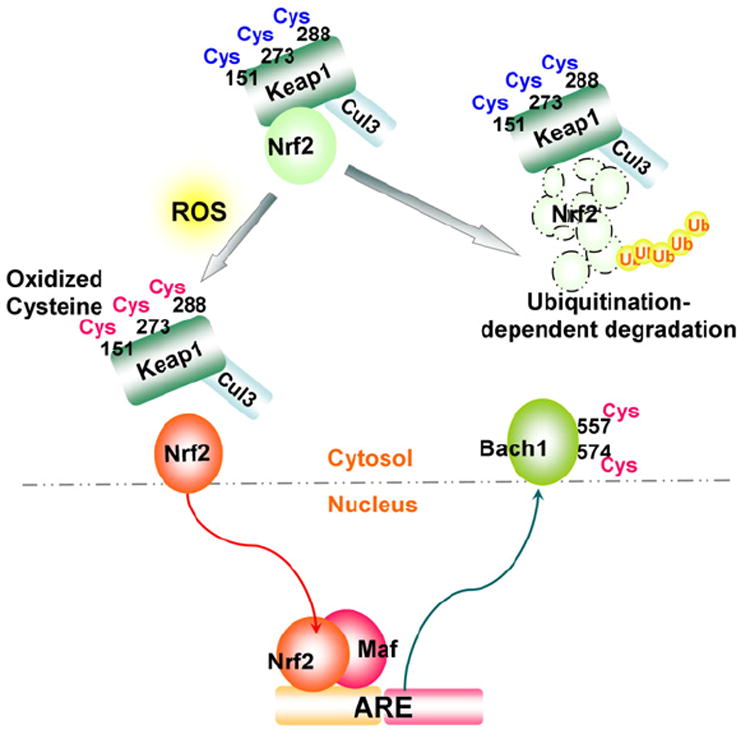
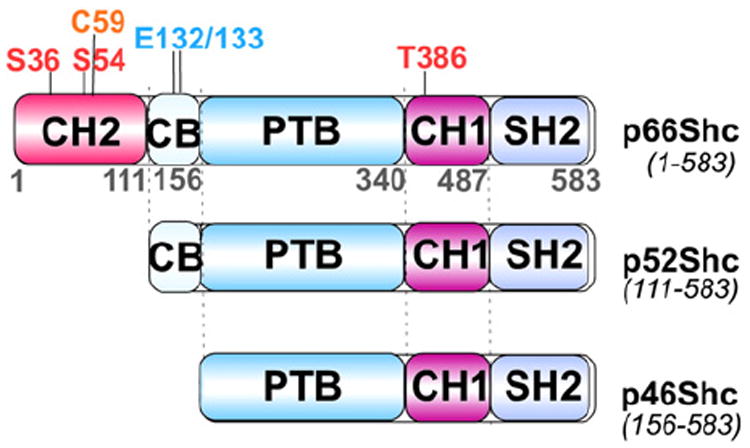
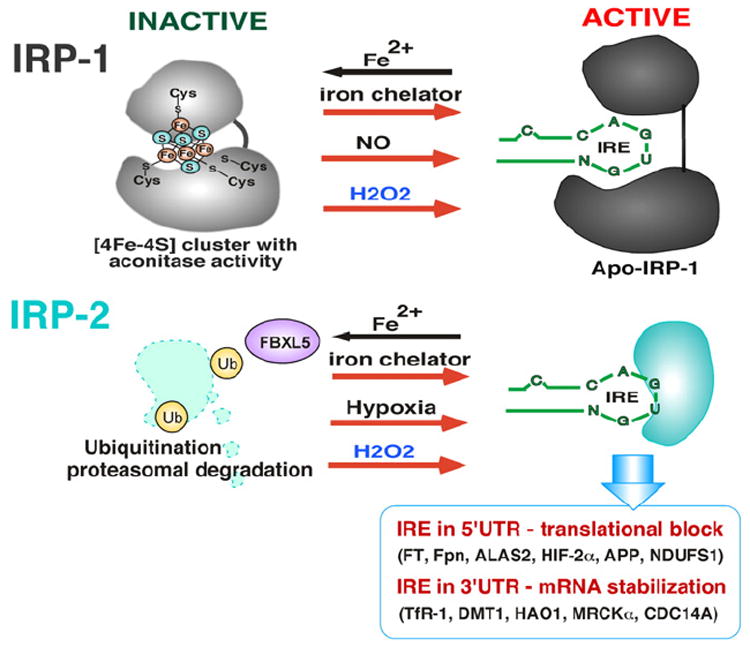
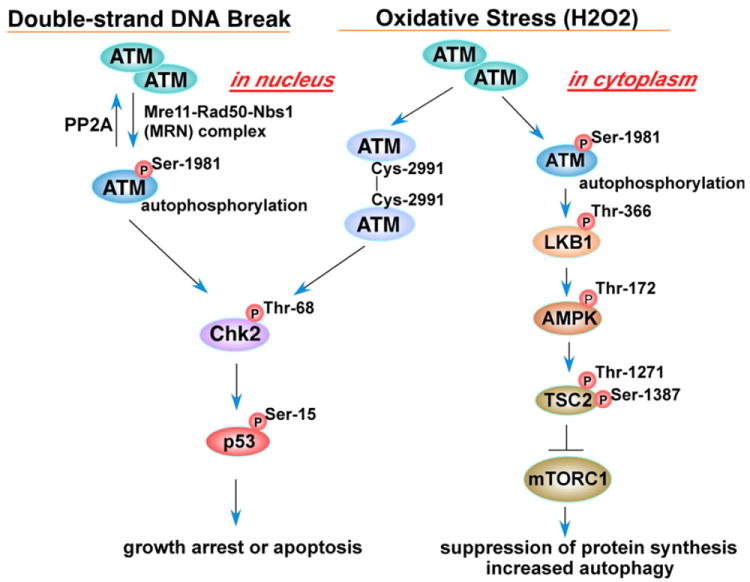
Reactive oxygen species (ROS) are generated during mitochondrial oxidative metabolism as well as in cellular response to xenobiotics, cytokines, and bacterial invasion. Oxidative stress refers to the imbalance due to excess ROS or oxidants over the capability of the cell to mount an effective antioxidant response. Oxidative stress results in macromolecular damage and is implicated in various disease states such as atherosclerosis, diabetes, cancer, neurodegeneration, and aging. Paradoxically, accumulating evidence indicates that ROS also serve as critical signaling molecules in cell proliferation and survival. While there is a large body of research demonstrating the general effect of oxidative stress on signaling pathways, less is known about the initial and direct regulation of signaling molecules by ROS, or what we term the "oxidative interface." Cellular ROS sensing and metabolism are tightly regulated by a variety of proteins involved in the redox (reduction/oxidation) mechanism. This review focuses on the molecular mechanisms through which ROS directly interact with critical signaling molecules to initiate signaling in a broad variety of cellular processes, such as proliferation and survival (MAP kinases, PI3 kinase, PTEN, and protein tyrosine phosphatases), ROS homeostasis and antioxidant gene regulation (thioredoxin, peroxiredoxin, Ref-1, and Nrf-2), mitochondrial oxidative stress, apoptosis, and aging (p66Shc), iron homeostasis through iron-sulfur cluster proteins (IRE-IRP), and ATM-regulated DNA damage response.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Oxidative stress and antioxidants in disease and cancer: a review.Asian Pac J Cancer Prev. 2014;15(11):4405-9. doi: 10.7314/apjcp.2014.15.11.4405. Asian Pac J Cancer Prev. 2014. PMID: 24969860 Review.
-
Oxidative stress and protein aggregation during biological aging.Exp Gerontol. 2001 Sep;36(9):1539-50. doi: 10.1016/s0531-5565(01)00139-5. Exp Gerontol. 2001. PMID: 11525876 Review.
-
Oxidized ATM promotes abnormal proliferation of breast CAFs through maintaining intracellular redox homeostasis and activating the PI3K-AKT, MEK-ERK, and Wnt-β-catenin signaling pathways.Cell Cycle. 2015;14(12):1908-24. doi: 10.1080/15384101.2015.1041685. Cell Cycle. 2015. PMID: 25970706 Free PMC article.
-
Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species.Redox Rep. 2014 Jan;19(1):8-15. doi: 10.1179/1351000213Y.0000000073. Epub 2013 Nov 22. Redox Rep. 2014. PMID: 24266943 Free PMC article. Review.
-
[Reactive Oxygen Species (ROS) Signaling: Regulatory Mechanisms and Pathophysiological Roles].Yakugaku Zasshi. 2019;139(10):1235-1241. doi: 10.1248/yakushi.19-00141. Yakugaku Zasshi. 2019. PMID: 31582606 Review. Japanese.
Cited by
-
Oxidative stress specifically downregulates survivin to promote breast tumour formation.Br J Cancer. 2013 Mar 5;108(4):848-58. doi: 10.1038/bjc.2013.40. Epub 2013 Feb 12. Br J Cancer. 2013. PMID: 23403820 Free PMC article.
-
The peroxisome counteracts oxidative stresses by suppressing catalase import via Pex14 phosphorylation.Elife. 2020 Aug 24;9:e55896. doi: 10.7554/eLife.55896. Elife. 2020. PMID: 32831175 Free PMC article.
-
Curcumin-mediated Photodynamic Therapy Inhibits the Phenotypic Transformation, Migration, and Foaming of Oxidized Low-density Lipoprotein-treated Vascular Smooth Muscle Cells by Promoting Autophagy.J Cardiovasc Pharmacol. 2021 Jun 2;78(2):308-318. doi: 10.1097/FJC.0000000000001069. J Cardiovasc Pharmacol. 2021. PMID: 34091481 Free PMC article.
-
An in-depth understanding of the role and mechanisms of T cells in immune organ aging and age-related diseases.Sci China Life Sci. 2025 Feb;68(2):328-353. doi: 10.1007/s11427-024-2695-x. Epub 2024 Sep 2. Sci China Life Sci. 2025. PMID: 39231902 Review.
-
DDX3 DEAD-box RNA helicase plays a central role in mitochondrial protein quality control in Leishmania.Cell Death Dis. 2016 Oct 13;7(10):e2406. doi: 10.1038/cddis.2016.315. Cell Death Dis. 2016. PMID: 27735940 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
