

Further evidence for the likely completeness of the library of solved single domain protein structures
- PMID: 22272723
- PMCID: PMC3351587
- DOI: 10.1021/jp211052j
Further evidence for the likely completeness of the library of solved single domain protein structures
Abstract
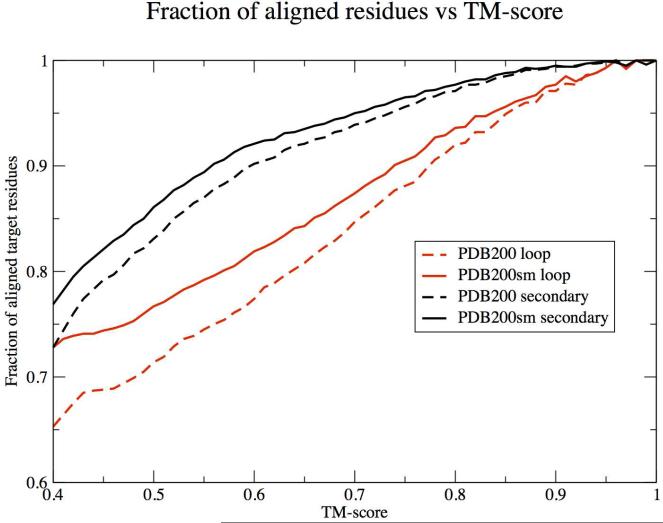
Recent studies questioned whether the Protein Data Bank (PDB) contains all compact, single domain protein structures. Here, we show that all quasi-spherical, QS, random protein structures devoid of secondary structure are in the PDB and are excellent templates for all native PDB proteins up to 250 residues. Because QS templates have a similar global contour as native, TASSER can refine 98% (90%) of those whose TM-score is 0.4 (0.35) to structures greater than or equal to the 0.5 TM-score threshold (0.74 (0.64) mean TM-score) for CATH/SCOP assignment. On the basis of this and the fact that, at a TM-score of 0.4, 83% (90%) of all (internal) core secondary structure elements are recovered, a 0.40 TM-score is an appropriate fold similarity assignment threshold. Despite the claims of Taylor, Trovato, and Zhou that many of their structures lack a PDB counterpart, using fr-TM-align, at a 0.45 (0.5) TM-score threshold, essentially all (most) are found in the PDB. Thus, the conclusion that the PDB is likely complete is further supported.
Figures









Similar articles
-
TM-align: a protein structure alignment algorithm based on the TM-score.Nucleic Acids Res. 2005 Apr 22;33(7):2302-9. doi: 10.1093/nar/gki524. Print 2005. Nucleic Acids Res. 2005. PMID: 15849316 Free PMC article.
-
Benchmarking of TASSER in the ab initio limit.Proteins. 2007 Jul 1;68(1):48-56. doi: 10.1002/prot.21392. Proteins. 2007. PMID: 17444524
-
The protein structure prediction problem could be solved using the current PDB library.Proc Natl Acad Sci U S A. 2005 Jan 25;102(4):1029-34. doi: 10.1073/pnas.0407152101. Epub 2005 Jan 14. Proc Natl Acad Sci U S A. 2005. PMID: 15653774 Free PMC article.
-
How significant is a protein structure similarity with TM-score = 0.5?Bioinformatics. 2010 Apr 1;26(7):889-95. doi: 10.1093/bioinformatics/btq066. Epub 2010 Feb 17. Bioinformatics. 2010. PMID: 20164152 Free PMC article.
-
TAPO: A combined method for the identification of tandem repeats in protein structures.FEBS Lett. 2015 Sep 14;589(19 Pt A):2611-9. doi: 10.1016/j.febslet.2015.08.025. Epub 2015 Aug 29. FEBS Lett. 2015. PMID: 26320412 Review.
Cited by
-
FINDSITE(comb): a threading/structure-based, proteomic-scale virtual ligand screening approach.J Chem Inf Model. 2013 Jan 28;53(1):230-40. doi: 10.1021/ci300510n. Epub 2012 Dec 28. J Chem Inf Model. 2013. PMID: 23240691 Free PMC article.
-
THE-DB: a threading model database for comparative protein structure analysis of the E. coli K12 and human proteomes.Database (Oxford). 2018 Jan 1;2018:bay090. doi: 10.1093/database/bay090. Database (Oxford). 2018. PMID: 30239678 Free PMC article.
-
EvoDesign: De novo protein design based on structural and evolutionary profiles.Nucleic Acids Res. 2013 Jul;41(Web Server issue):W273-80. doi: 10.1093/nar/gkt384. Epub 2013 May 13. Nucleic Acids Res. 2013. PMID: 23671331 Free PMC article.
-
Protein folding and de novo protein design for biotechnological applications.Trends Biotechnol. 2014 Feb;32(2):99-109. doi: 10.1016/j.tibtech.2013.10.008. Epub 2013 Nov 19. Trends Biotechnol. 2014. PMID: 24268901 Free PMC article. Review.
-
A review of visualisations of protein fold networks and their relationship with sequence and function.Biol Rev Camb Philos Soc. 2023 Feb;98(1):243-262. doi: 10.1111/brv.12905. Epub 2022 Oct 9. Biol Rev Camb Philos Soc. 2023. PMID: 36210328 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
