

Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT
- PMID: 22245495
- PMCID: PMC3298765
- DOI: 10.1016/j.cellsig.2011.12.024
Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT
Abstract
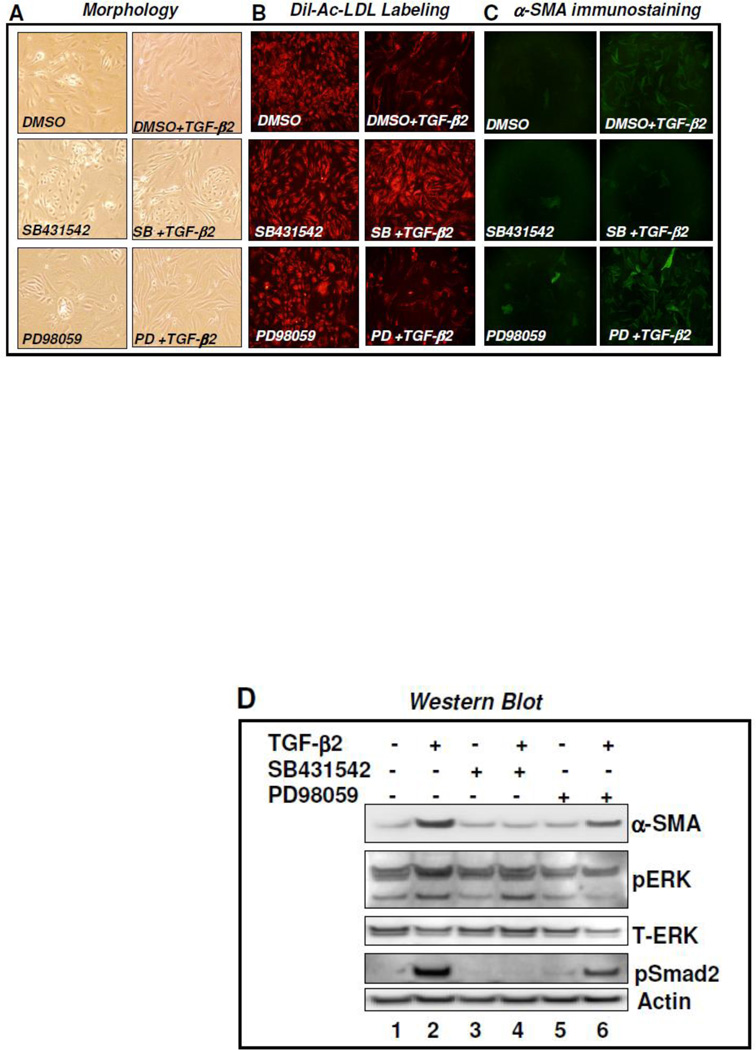
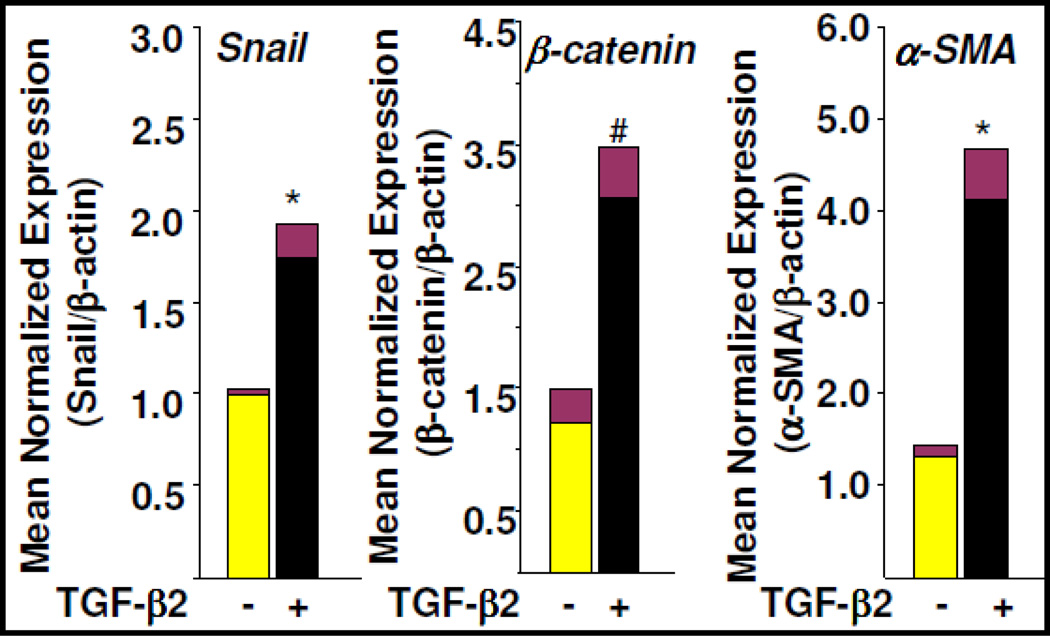
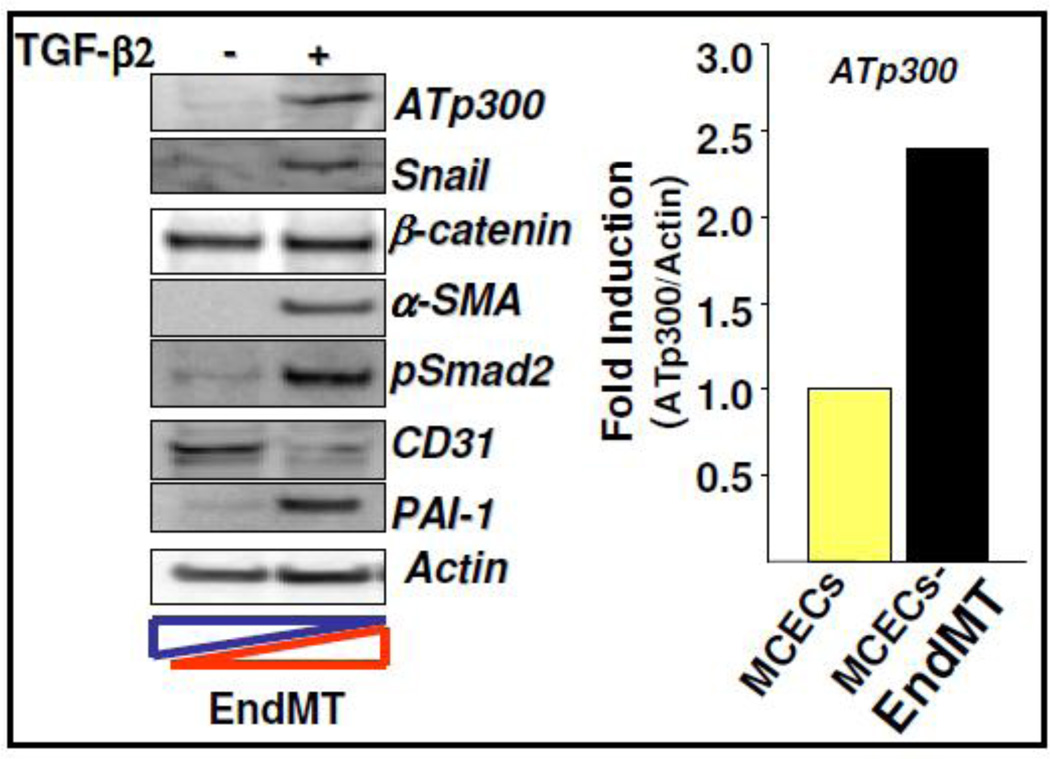
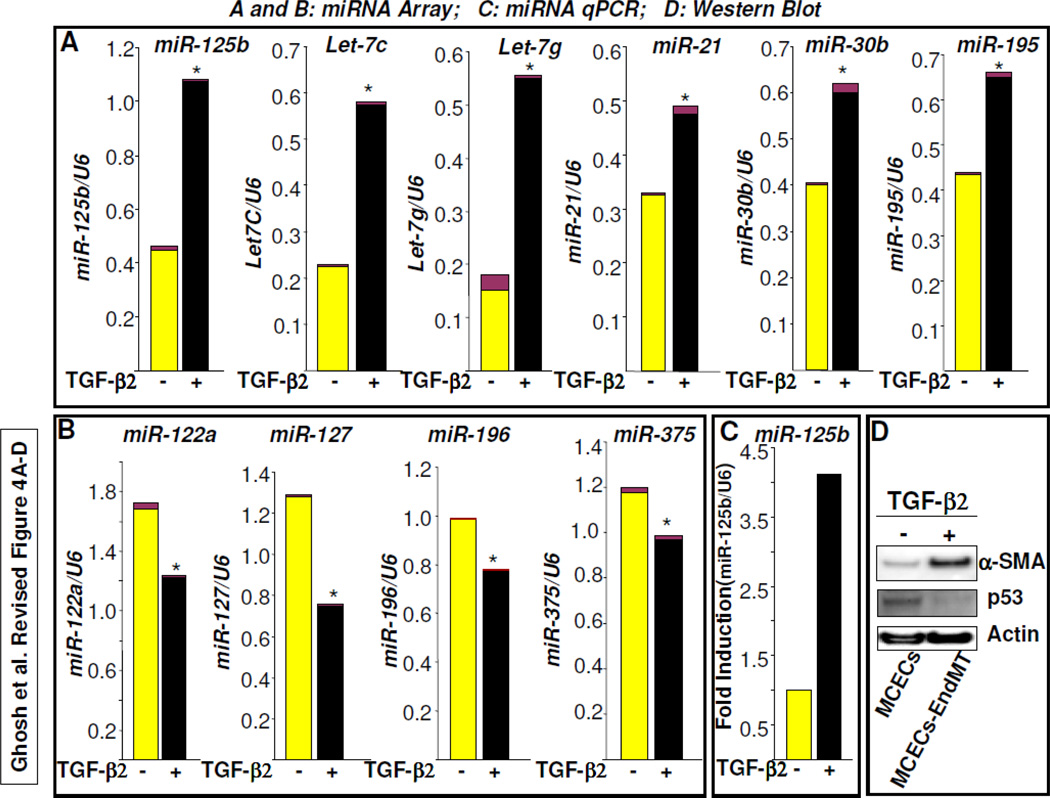
Fibroblasts are responsible for producing the majority of collagen and other extracellular matrix (ECM) proteins in tissues. In the injured tissue, transforming growth factor-β (TGF-β)-activated fibroblasts or differentiated myofibroblasts synthesize excessive ECM proteins and play a pivotal role in the pathogenesis of fibrosis in heart, kidney and other organs. Recent studies suggest that fibroblast-like cells, derived from endothelial cells by endothelial-to-mesenchymal transition (EndMT), contribute to the pathogenesis of cardiac fibrosis. The molecular basis of EndMT, however, is poorly understood. Here, we investigated the molecular basis of EndMT in mouse cardiac endothelial cells (MCECs) in response to TGF-β2. MCECs exposed to TGF-β2 underwent EndMT as evidenced by morphologic changes, lack of acetylated-low density lipoprotein (Ac-LDL) uptake, and the presence of alpha-smooth muscle actin (α-SMA) staining. Treatment with SB431542, a small molecule inhibitor of TGF-β-receptor I (TβRI) kinase, but not PD98059, a MEK inhibitor, completely blocked TGF-β2-induced EndMT. The transcript and protein levels of α-SMA, Snail and β-catenin as well as acetyltransferase p300 (ATp300) were elevated in EndMT derived fibroblast-like cells. Importantly, microRNA (miRNA) array data revealed that the expression levels of specific miRNAs, known to be dysregulated in different cardiovascular diseases, were altered during EndMT. The protein level of cellular p53, a bonafide target of miR-125b, was downregulated in EndMT-derived fibroblast-like cells. Here, we report for the first time, the differential expression of miRNAs during cardiac EndMT. These results collectively suggest that TβRI serine-threonine kinase-induced TGF-β signaling and microRNAs, the epigenetic regulator of gene expression at the posttranscriptional level, are involved in EndMT and promote profibrotic signaling in EndMT-derived fibroblast-like cells. Pharmacologic agents that restrict the progression of cardiac EndMT, a phenomenon that is found in adults only in the pathological conditions, in targeting specific miRNA may be helpful in preventing and treating cardiac fibrosis.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
The mechanism of TGF-β/miR-155/c-Ski regulates endothelial-mesenchymal transition in human coronary artery endothelial cells.Biosci Rep. 2017 Aug 23;37(4):BSR20160603. doi: 10.1042/BSR20160603. Print 2017 Aug 31. Biosci Rep. 2017. PMID: 28607031 Free PMC article.
-
Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis.J Mol Cell Cardiol. 2019 Feb;127:83-96. doi: 10.1016/j.yjmcc.2018.12.002. Epub 2018 Dec 7. J Mol Cell Cardiol. 2019. PMID: 30529267
-
Crosstalk between endothelial cell-specific calpain inhibition and the endothelial-mesenchymal transition via the HSP90/Akt signaling pathway.Biomed Pharmacother. 2020 Apr;124:109822. doi: 10.1016/j.biopha.2020.109822. Epub 2020 Jan 17. Biomed Pharmacother. 2020. PMID: 31958767
-
Transcriptional regulation of endothelial-to-mesenchymal transition in cardiac fibrosis: role of myocardin-related transcription factor A and activating transcription factor 3.Can J Physiol Pharmacol. 2017 Oct;95(10):1263-1270. doi: 10.1139/cjpp-2016-0634. Epub 2017 Jul 7. Can J Physiol Pharmacol. 2017. PMID: 28686848 Review.
-
Regulation of endothelial cell plasticity by TGF-β.Cell Tissue Res. 2012 Jan;347(1):177-86. doi: 10.1007/s00441-011-1222-6. Epub 2011 Aug 25. Cell Tissue Res. 2012. PMID: 21866313 Free PMC article. Review.
Cited by
-
Astragalus Mongholicus: A review of its anti-fibrosis properties.Front Pharmacol. 2022 Sep 7;13:976561. doi: 10.3389/fphar.2022.976561. eCollection 2022. Front Pharmacol. 2022. PMID: 36160396 Free PMC article. Review.
-
Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review.J Am Coll Cardiol. 2019 Jan 22;73(2):190-209. doi: 10.1016/j.jacc.2018.09.089. J Am Coll Cardiol. 2019. PMID: 30654892 Free PMC article. Review.
-
Let-7 in cardiovascular diseases, heart development and cardiovascular differentiation from stem cells.Int J Mol Sci. 2013 Nov 21;14(11):23086-102. doi: 10.3390/ijms141123086. Int J Mol Sci. 2013. PMID: 24284400 Free PMC article. Review.
-
Endothelial-to-Mesenchymal Transition in Cardiovascular Pathophysiology.Int J Mol Sci. 2024 Jun 4;25(11):6180. doi: 10.3390/ijms25116180. Int J Mol Sci. 2024. PMID: 38892367 Free PMC article. Review.
-
Plasminogen Activator Inhibitor Type I Controls Cardiomyocyte Transforming Growth Factor-β and Cardiac Fibrosis.Circulation. 2017 Aug 15;136(7):664-679. doi: 10.1161/CIRCULATIONAHA.117.028145. Epub 2017 Jun 6. Circulation. 2017. PMID: 28588076 Free PMC article.
References
-
- Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;293:L1–L8. - PubMed
-
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Nat. Med. 2007;13:952–961. - PubMed
-
- Goumans MJ, van Zonneveld AJ, ten Dijke P. Trends Cardiovasc. Med. 2008;18:293–298. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
