

The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation
- PMID: 22195744
- PMCID: PMC3248798
- DOI: 10.1016/j.immuni.2011.09.021
The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation
Abstract
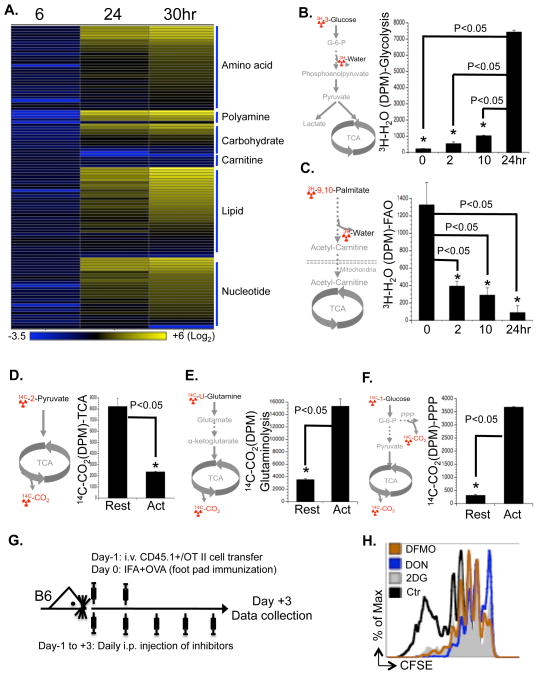
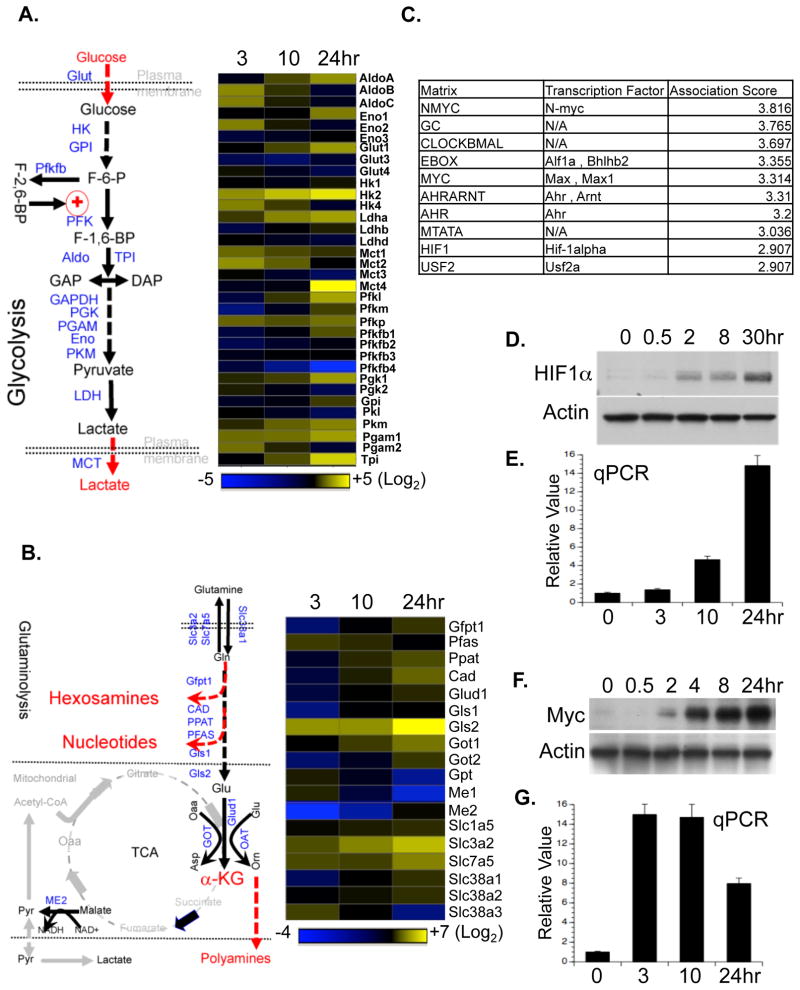
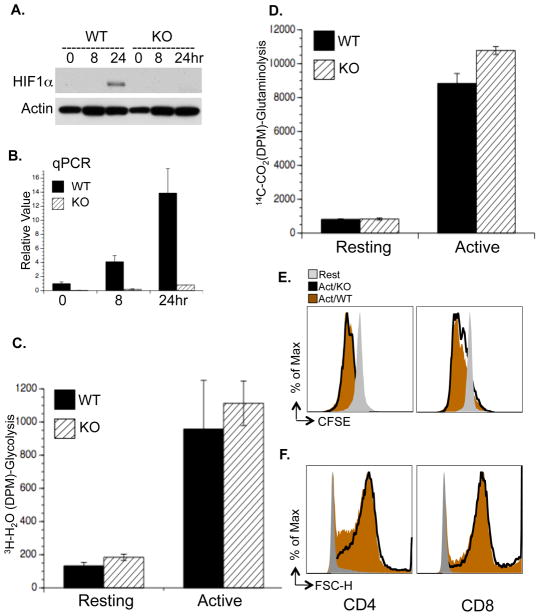
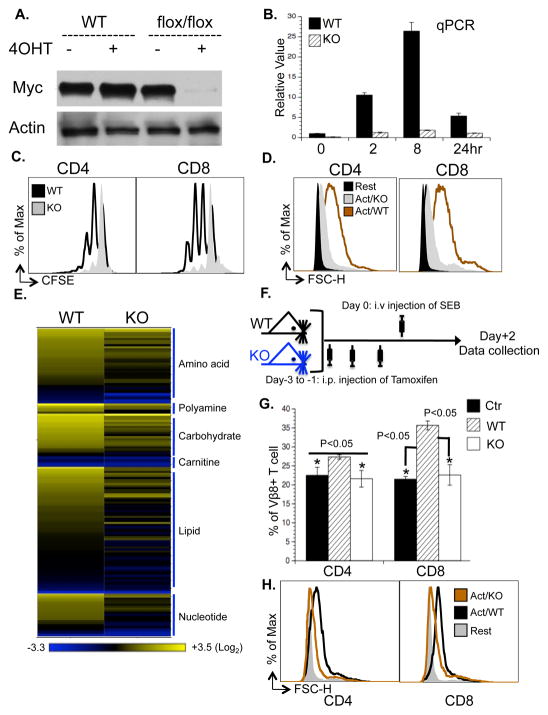
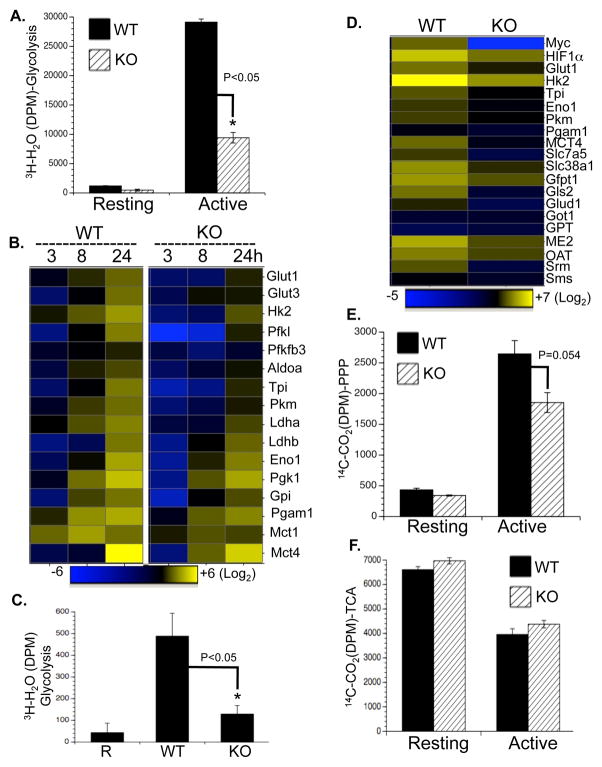
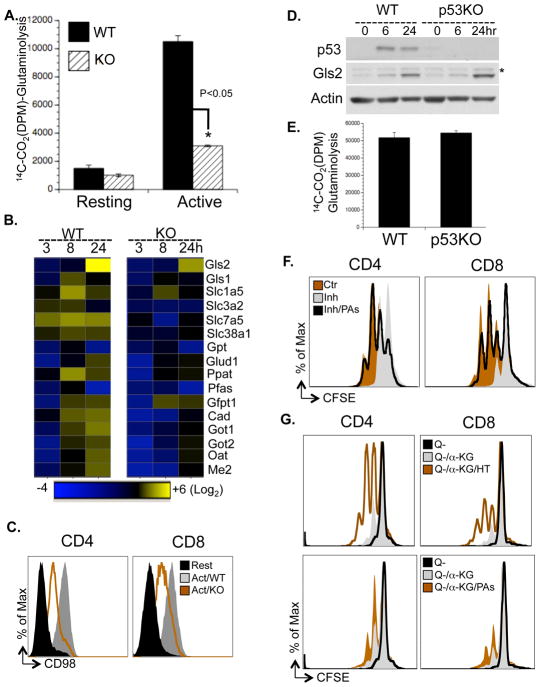
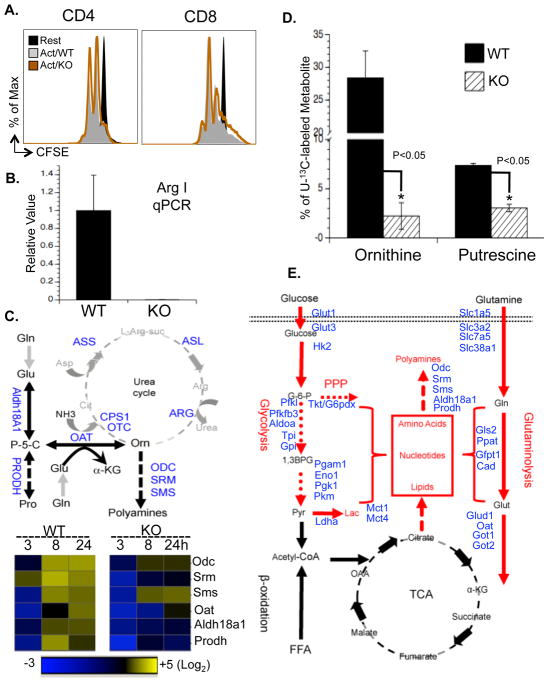
To fulfill the bioenergetic and biosynthetic demand of proliferation, T cells reprogram their metabolic pathways from fatty acid β-oxidation and pyruvate oxidation via the TCA cycle to the glycolytic, pentose-phosphate, and glutaminolytic pathways. Two of the top-ranked candidate transcription factors potentially responsible for the activation-induced T cell metabolic transcriptome, HIF1α and Myc, were induced upon T cell activation, but only the acute deletion of Myc markedly inhibited activation-induced glycolysis and glutaminolysis in T cells. Glutamine deprivation compromised activation-induced T cell growth and proliferation, and this was partially replaced by nucleotides and polyamines, implicating glutamine as an important source for biosynthetic precursors in active T cells. Metabolic tracer analysis revealed a Myc-dependent metabolic pathway linking glutaminolysis to the biosynthesis of polyamines. Therefore, a Myc-dependent global metabolic transcriptome drives metabolic reprogramming in activated, primary T lymphocytes. This may represent a general mechanism for metabolic reprogramming under patho-physiological conditions.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Comment in
-
T cell Myc-tabolism.Immunity. 2011 Dec 23;35(6):845-6. doi: 10.1016/j.immuni.2011.12.001. Immunity. 2011. PMID: 22195738 Free PMC article.
Similar articles
-
Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent.Proc Natl Acad Sci U S A. 2016 Feb 9;113(6):1564-9. doi: 10.1073/pnas.1518000113. Epub 2016 Jan 25. Proc Natl Acad Sci U S A. 2016. PMID: 26811453 Free PMC article.
-
MYC and HIF in shaping immune response and immune metabolism.Cytokine Growth Factor Rev. 2017 Jun;35:63-70. doi: 10.1016/j.cytogfr.2017.03.004. Epub 2017 Mar 23. Cytokine Growth Factor Rev. 2017. PMID: 28363691 Review.
-
Inhibitor of differentiation 1 transcription factor promotes metabolic reprogramming in hepatocellular carcinoma cells.FASEB J. 2016 Jan;30(1):262-75. doi: 10.1096/fj.15-277749. Epub 2015 Sep 1. FASEB J. 2016. PMID: 26330493 Free PMC article.
-
Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression.Oncotarget. 2015 Sep 22;6(28):26161-76. doi: 10.18632/oncotarget.4544. Oncotarget. 2015. PMID: 26317652 Free PMC article.
-
MYC and tumor metabolism: chicken and egg.EMBO J. 2017 Dec 1;36(23):3409-3420. doi: 10.15252/embj.201796438. Epub 2017 Nov 10. EMBO J. 2017. PMID: 29127156 Free PMC article. Review.
Cited by
-
Deep learning analysis of histopathological images predicts immunotherapy prognosis and reveals tumour microenvironment features in non-small cell lung cancer.Br J Cancer. 2024 Dec;131(11):1833-1845. doi: 10.1038/s41416-024-02856-8. Epub 2024 Oct 25. Br J Cancer. 2024. PMID: 39455880
-
Critical Role of AdipoR1 in Regulating Th17 Cell Differentiation Through Modulation of HIF-1α-Dependent Glycolysis.Front Immunol. 2020 Aug 18;11:2040. doi: 10.3389/fimmu.2020.02040. eCollection 2020. Front Immunol. 2020. PMID: 32973810 Free PMC article.
-
Famine versus feast: understanding the metabolism of tumors in vivo.Trends Biochem Sci. 2015 Mar;40(3):130-40. doi: 10.1016/j.tibs.2015.01.004. Epub 2015 Jan 29. Trends Biochem Sci. 2015. PMID: 25639751 Free PMC article. Review.
-
RNA Flow Cytometry for the Study of T Cell Metabolism.Int J Mol Sci. 2021 Apr 9;22(8):3906. doi: 10.3390/ijms22083906. Int J Mol Sci. 2021. PMID: 33918901 Free PMC article. Review.
-
L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity.Cell. 2016 Oct 20;167(3):829-842.e13. doi: 10.1016/j.cell.2016.09.031. Epub 2016 Oct 13. Cell. 2016. PMID: 27745970 Free PMC article.
References
-
- Bowlin TL, McKown BJ, Babcock GF, Sunkara PS. Intracellular polyamine biosynthesis is required for interleukin 2 responsiveness during lymphocyte mitogenesis. Cell Immunol. 1987;106:420–427. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 GM052735-21/GM/NIGMS NIH HHS/United States
- R56 AI081773/AI/NIAID NIH HHS/United States
- R37 GM052735/GM/NIGMS NIH HHS/United States
- AI40646/AI/NIAID NIH HHS/United States
- S064599/PHS HHS/United States
- R01 AI040646/AI/NIAID NIH HHS/United States
- R01 GM052735/GM/NIGMS NIH HHS/United States
- R01 AI081773/AI/NIAID NIH HHS/United States
- GM52735/GM/NIGMS NIH HHS/United States
- K01 AR053573/AR/NIAMS NIH HHS/United States
- AR053573/AR/NIAMS NIH HHS/United States
- AI081773/AI/NIAID NIH HHS/United States
- R01 AI040646-16/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
