

Protein conformational dynamics in the mechanism of HIV-1 protease catalysis
- PMID: 22158985
- PMCID: PMC3248522
- DOI: 10.1073/pnas.1111202108
Protein conformational dynamics in the mechanism of HIV-1 protease catalysis
Abstract
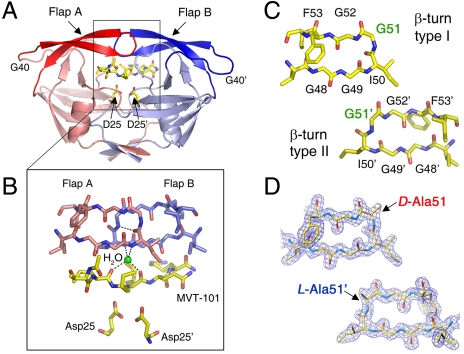
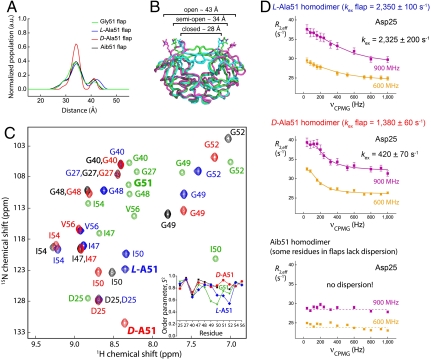
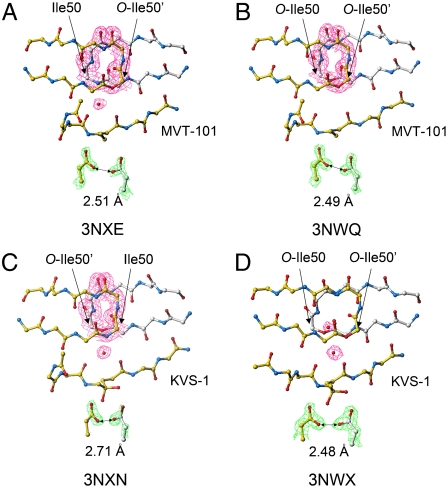
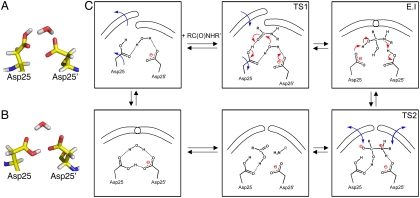
We have used chemical protein synthesis and advanced physical methods to probe dynamics-function correlations for the HIV-1 protease, an enzyme that has received considerable attention as a target for the treatment of AIDS. Chemical synthesis was used to prepare a series of unique analogues of the HIV-1 protease in which the flexibility of the "flap" structures (residues 37-61 in each monomer of the homodimeric protein molecule) was systematically varied. These analogue enzymes were further studied by X-ray crystallography, NMR relaxation, and pulse-EPR methods, in conjunction with molecular dynamics simulations. We show that conformational isomerization in the flaps is correlated with structural reorganization of residues in the active site, and that it is preorganization of the active site that is a rate-limiting factor in catalysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Flexibility and function in HIV-1 protease.Nat Struct Biol. 1995 Apr;2(4):274-80. doi: 10.1038/nsb0495-274. Nat Struct Biol. 1995. PMID: 7796263
-
Probing the S1/S1' substrate binding pocket geometry of HIV-1 protease with modified aspartic acid analogues.Biochemistry. 2000 Aug 1;39(30):8768-81. doi: 10.1021/bi000214t. Biochemistry. 2000. PMID: 10913288
-
Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance.Structure. 2000 Dec 15;8(12):1259-65. doi: 10.1016/s0969-2126(00)00537-2. Structure. 2000. PMID: 11188690
-
Targeting structural flexibility in HIV-1 protease inhibitor binding.Drug Discov Today. 2007 Feb;12(3-4):132-8. doi: 10.1016/j.drudis.2006.12.011. Epub 2006 Dec 20. Drug Discov Today. 2007. PMID: 17275733 Free PMC article. Review.
-
Catalytic contributions from remote regions of enzyme structure.Chem Rev. 2011 Dec 14;111(12):7595-624. doi: 10.1021/cr100042n. Epub 2011 Sep 19. Chem Rev. 2011. PMID: 21923192 Review. No abstract available.
Cited by
-
Comprehending the Structure, Dynamics, and Mechanism of Action of Drug-Resistant HIV Protease.ACS Omega. 2023 Mar 7;8(11):9748-9763. doi: 10.1021/acsomega.2c08279. eCollection 2023 Mar 21. ACS Omega. 2023. PMID: 36969469 Free PMC article. Review.
-
Comparison of design strategies for α-helix backbone modification in a protein tertiary fold.Chem Commun (Camb). 2016 Mar 7;52(19):3789-92. doi: 10.1039/c6cc00273k. Chem Commun (Camb). 2016. PMID: 26853882 Free PMC article.
-
Role of Conformational Motions in Enzyme Function: Selected Methodologies and Case Studies.Catalysts. 2016 Jun;6(6):81. doi: 10.3390/catal6060081. Epub 2016 May 27. Catalysts. 2016. PMID: 28367322 Free PMC article.
-
A molecular engineering toolbox for the structural biologist.Q Rev Biophys. 2017 Jan;50:e7. doi: 10.1017/S0033583517000051. Q Rev Biophys. 2017. PMID: 29233219 Free PMC article. Review.
-
The role of select subtype polymorphisms on HIV-1 protease conformational sampling and dynamics.J Biol Chem. 2014 Jun 13;289(24):17203-14. doi: 10.1074/jbc.M114.571836. Epub 2014 Apr 17. J Biol Chem. 2014. PMID: 24742668 Free PMC article.
References
-
- Pomerantz RJ, Horn DL. Twenty years of therapy for HIV-1 infection. Nat Med. 2003;9:867–873. - PubMed
-
- Nicholson LK, et al. Flexibility and function in HIV-1 protease. Nat Struct Biol. 1995;2:274–280. - PubMed
-
- Ishima R, Louis JM. A diverse view of protein dynamics from NMR studies of HIV-1 protease flaps. Proteins. 2008;70:1408–1415. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
