

Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency
- PMID: 22105173
- PMCID: PMC3226004
- DOI: 10.1172/JCI59259
Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency
Abstract
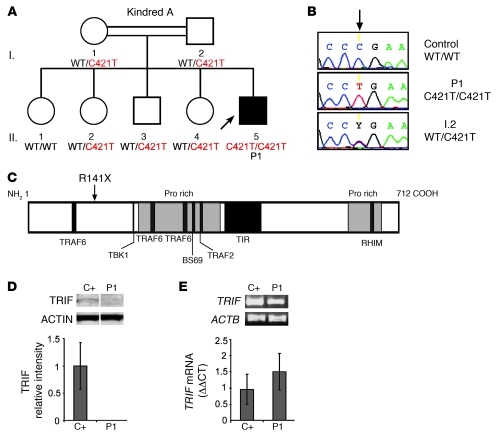
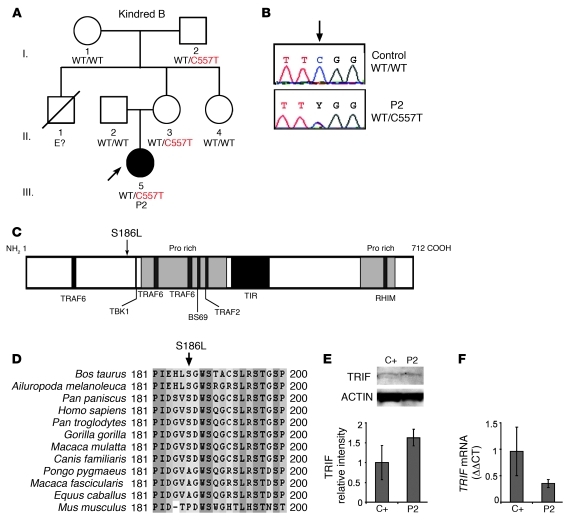
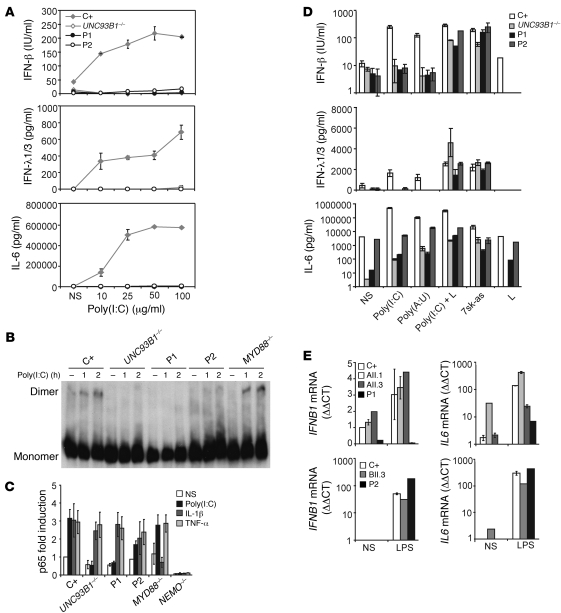
Herpes simplex encephalitis (HSE) is the most common sporadic viral encephalitis of childhood. Autosomal recessive (AR) UNC-93B and TLR3 deficiencies and autosomal dominant (AD) TLR3 and TRAF3 deficiencies underlie HSE in some children. We report here unrelated HSE children with AR or AD TRIF deficiency. The AR form of the disease was found to be due to a homozygous nonsense mutation that resulted in a complete absence of the TRIF protein. Both the TLR3- and the TRIF-dependent TLR4 signaling pathways were abolished. The AD form of disease was found to be due to a heterozygous missense mutation, resulting in a dysfunctional protein. In this form of the disease, the TLR3 signaling pathway was impaired, whereas the TRIF-dependent TLR4 pathway was unaffected. Both patients, however, showed reduced capacity to respond to stimulation of the DExD/H-box helicases pathway. To date, the TRIF-deficient patients with HSE described herein have suffered from no other infections. Moreover, as observed in patients with other genetic etiologies of HSE, clinical penetrance was found to be incomplete, as some HSV-1-infected TRIF-deficient relatives have not developed HSE. Our results provide what we believe to be the first description of human TRIF deficiency and a new genetic etiology for HSE. They suggest that the TRIF-dependent TLR4 and DExD/H-box helicase pathways are largely redundant in host defense. They further demonstrate the importance of TRIF for the TLR3-dependent production of antiviral IFNs in the CNS during primary infection with HSV-1 in childhood.
Figures



Similar articles
-
TLR3/TRIF pathway confers protection against herpes simplex encephalitis through NK cell activation mediated by a loop of type I IFN and IL-15 from epithelial and dendritic cells.Immunology. 2023 Sep;170(1):83-104. doi: 10.1111/imm.13664. Epub 2023 Jun 5. Immunology. 2023. PMID: 37278103
-
TLR3 deficiency in herpes simplex encephalitis: high allelic heterogeneity and recurrence risk.Neurology. 2014 Nov 18;83(21):1888-97. doi: 10.1212/WNL.0000000000000999. Epub 2014 Oct 22. Neurology. 2014. PMID: 25339207 Free PMC article.
-
Mendelian predisposition to herpes simplex encephalitis.Handb Clin Neurol. 2013;112:1091-7. doi: 10.1016/B978-0-444-52910-7.00027-1. Handb Clin Neurol. 2013. PMID: 23622315 Review.
-
Herpes simplex virus encephalitis in human UNC-93B deficiency.Science. 2006 Oct 13;314(5797):308-12. doi: 10.1126/science.1128346. Epub 2006 Sep 14. Science. 2006. PMID: 16973841
-
Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity.Hum Genet. 2020 Jun;139(6-7):911-918. doi: 10.1007/s00439-020-02127-5. Epub 2020 Feb 10. Hum Genet. 2020. PMID: 32040615 Free PMC article. Review.
Cited by
-
Human TMEFF1 is a restriction factor for herpes simplex virus in the brain.Nature. 2024 Aug;632(8024):390-400. doi: 10.1038/s41586-024-07745-x. Epub 2024 Jul 24. Nature. 2024. PMID: 39048830 Free PMC article.
-
Orchestration of antiviral responses within the infected central nervous system.Cell Mol Immunol. 2024 Sep;21(9):943-958. doi: 10.1038/s41423-024-01181-7. Epub 2024 Jul 12. Cell Mol Immunol. 2024. PMID: 38997413 Free PMC article. Review.
-
Herpes simplex encephalitis due to a mutation in an E3 ubiquitin ligase.Nat Commun. 2024 May 10;15(1):3969. doi: 10.1038/s41467-024-48287-0. Nat Commun. 2024. PMID: 38730242 Free PMC article.
-
Whole exome sequencing of patients with varicella-zoster virus and herpes simplex virus induced acute retinal necrosis reveals rare disease-associated genetic variants.Front Mol Neurosci. 2023 Oct 25;16:1253040. doi: 10.3389/fnmol.2023.1253040. eCollection 2023. Front Mol Neurosci. 2023. PMID: 38025266 Free PMC article.
-
An IKBKE variant conferring functional cGAS/STING pathway deficiency and susceptibility to recurrent HSV-2 meningitis.JCI Insight. 2023 Nov 8;8(21):e173066. doi: 10.1172/jci.insight.173066. JCI Insight. 2023. PMID: 37937644 Free PMC article.
References
-
- Gordon B, Selnes OA, Hart J, Jr, Hanley DF, Whitley RJ. Long-term cognitive sequelae of acyclovir–treated herpes simplex encephalitis. Arch Neurol. 1990;47(6):646–647. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
