

A noncanonical Flt3ITD/NF-κB signaling pathway represses DAPK1 in acute myeloid leukemia
- PMID: 22096027
- PMCID: PMC3918433
- DOI: 10.1158/1078-0432.CCR-10-3022
A noncanonical Flt3ITD/NF-κB signaling pathway represses DAPK1 in acute myeloid leukemia
Abstract
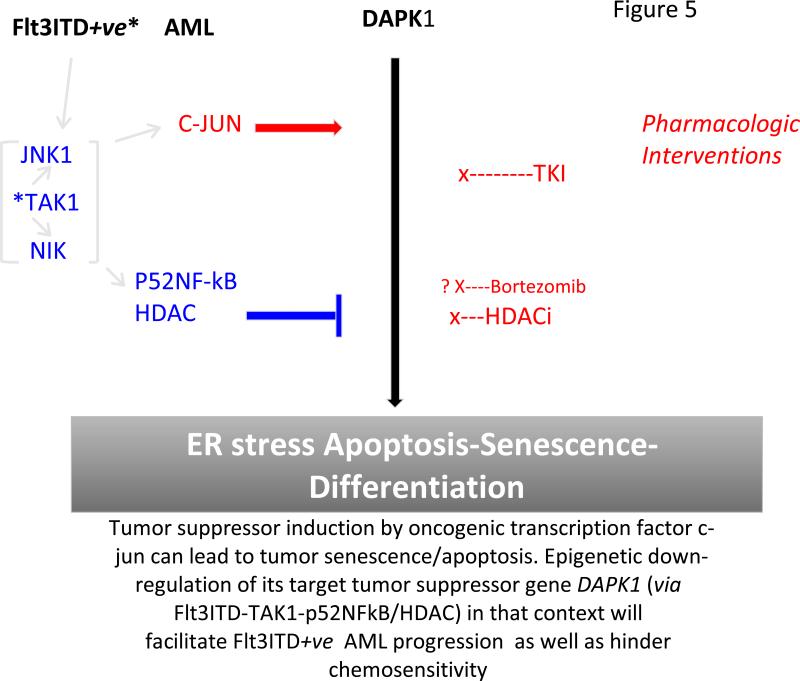
Purpose: Death-associated protein kinase 1 (DAPK1), a tumor suppressor, is a rate-limiting effector in an endoplasmic reticulum (ER) stress-dependent apoptotic pathway. Its expression is epigenetically suppressed in several tumors. A mechanistic basis for epigenetic/transcriptional repression of DAPK1 was investigated in certain forms of acute myeloid leukemia (AML) with poor prognosis, which lacked ER stress-induced apoptosis.
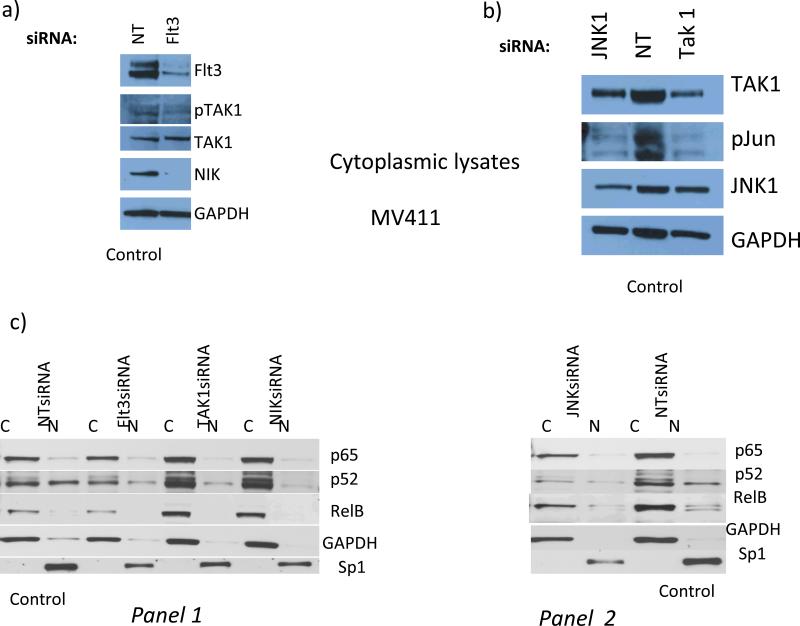
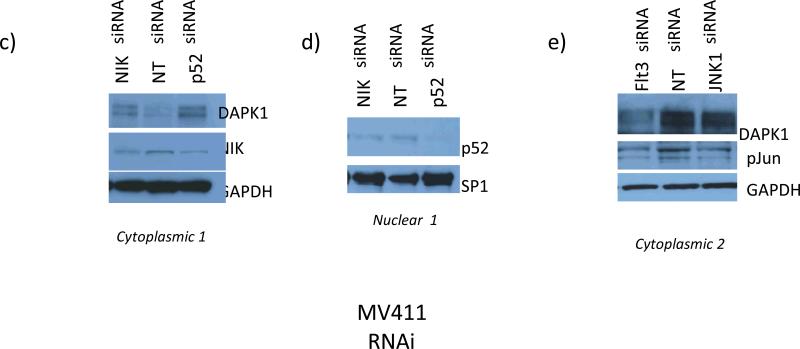
Experimental design: Heterogeneous primary AMLs were screened to identify a subgroup with Flt3ITD in which repression of DAPK1, among NF-κB-and c-Jun-responsive genes, was studied. RNA interference knockdown studies were carried out in an Flt3ITD(+) cell line, MV-4-11, to establish genetic epistasis in the pathway Flt3ITD-TAK1-DAPK1 repression, and chromatin immunoprecipitations were carried out to identify proximate effector proteins, including TAK1-activated p52NF-κB, at the DAPK1 locus.
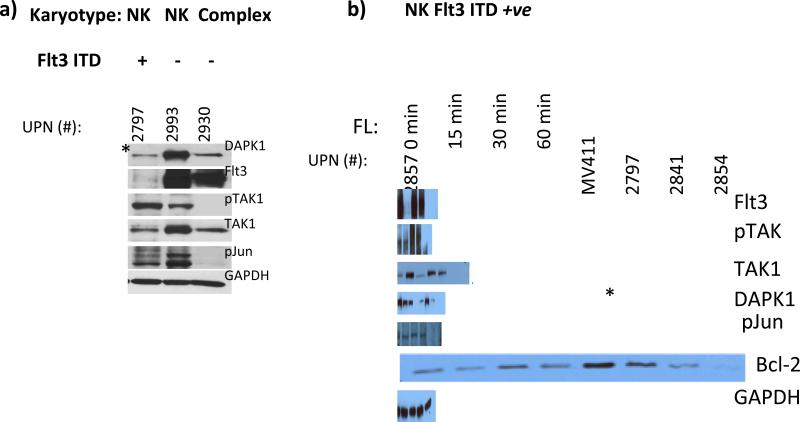
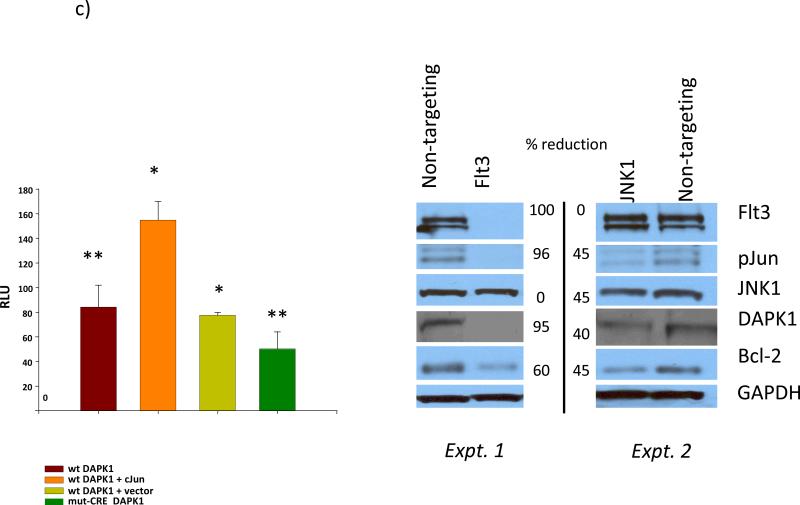
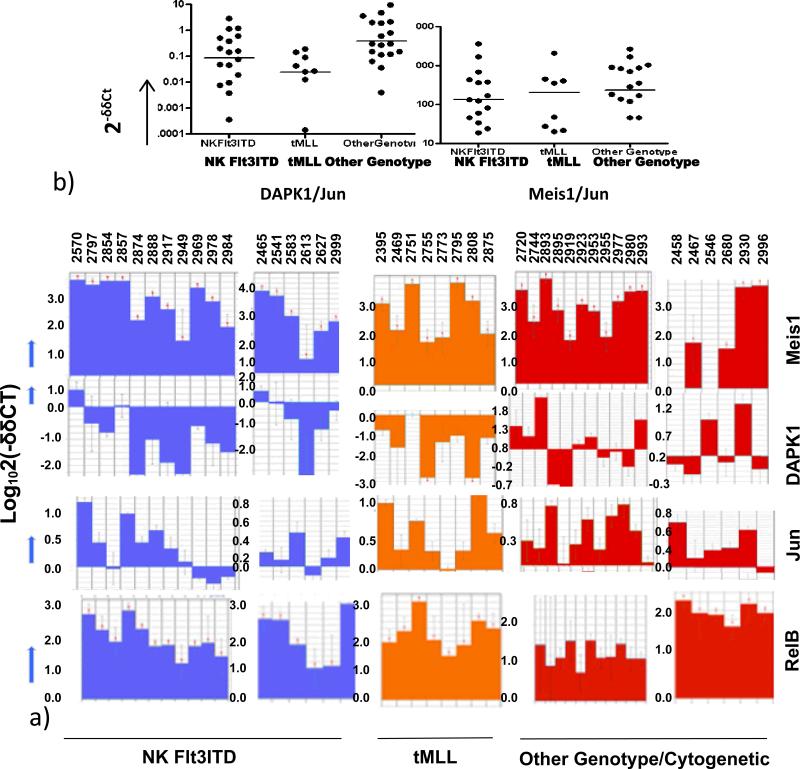
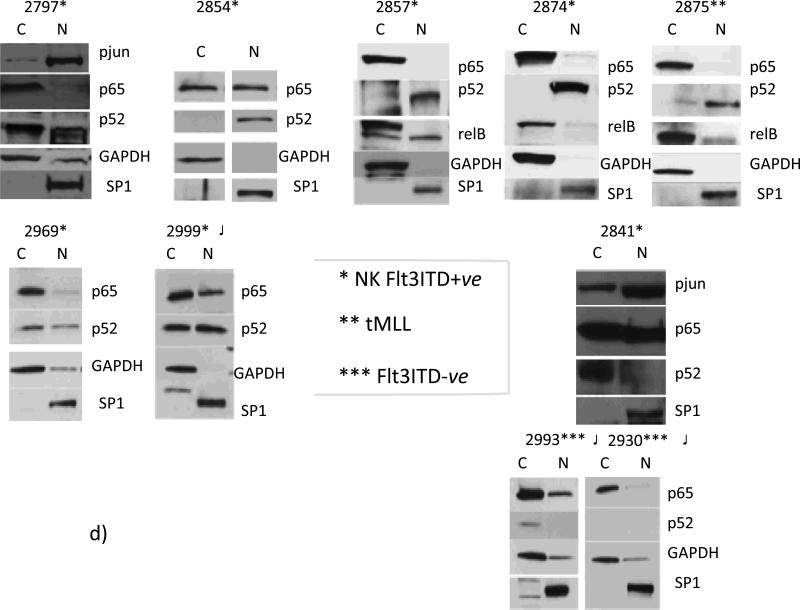
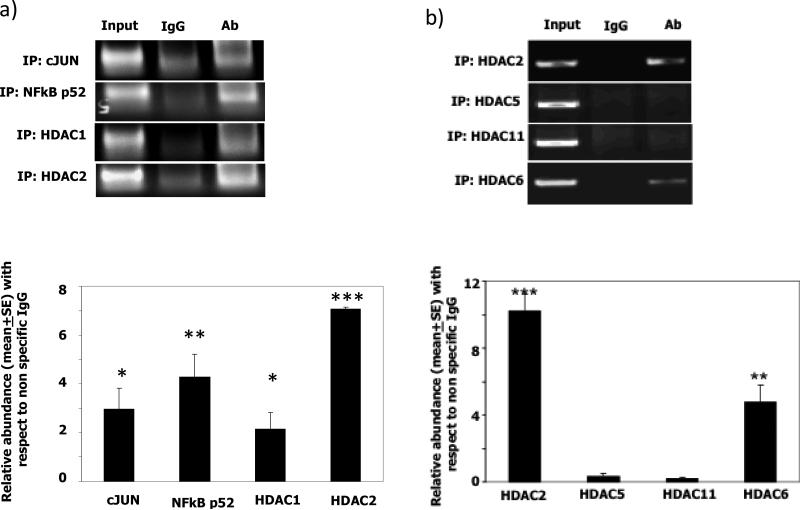
Results: AMLs characterized by normal karyotype with Flt3ITD were found to have 10- to 100-fold lower DAPK1 transcripts normalized to the expression of c-Jun, a transcriptional activator of DAPK1, as compared with a heterogeneous cytogenetic category. In addition, Meis1, a c-Jun-responsive adverse AML prognostic gene signature was measured as control. These Flt3ITD(+) AMLs overexpress relB, a transcriptional repressor, which forms active heterodimers with p52NF-κB. Chromatin immunoprecipitation assays identified p52NF-κB binding to the DAPK1 promoter together with histone deacetylase 2 (HDAC2) and HDAC6 in the Flt3ITD(+) human AML cell line MV-4-11. Knockdown of p52NF-κB or its upstream regulator, NF-κB-inducing kinase (NIK), de-repressed DAPK1. DAPK1-repressed primary Flt3ITD(+) AMLs had selective nuclear activation of p52NF-κB.
Conclusions: Flt3ITD promotes a noncanonical pathway via TAK1 and p52NF-κB to suppress DAPK1 in association with HDACs, which explains DAPK1 repression in Flt3ITD(+) AML.
©2011 AACR.
Figures
Similar articles
-
Synergistic killing of FLT3ITD-positive AML cells by combined inhibition of tyrosine-kinase activity and N-glycosylation.Oncotarget. 2017 Apr 18;8(16):26613-26624. doi: 10.18632/oncotarget.15772. Oncotarget. 2017. PMID: 28460451 Free PMC article.
-
Critical role for transcription factor C/EBP-beta in regulating the expression of death-associated protein kinase 1.Mol Cell Biol. 2008 Apr;28(8):2528-48. doi: 10.1128/MCB.00784-07. Epub 2008 Feb 4. Mol Cell Biol. 2008. PMID: 18250155 Free PMC article.
-
DAPk1 inhibits NF-κB activation through TNF-α and INF-γ-induced apoptosis.Cell Signal. 2012 Jul;24(7):1471-7. doi: 10.1016/j.cellsig.2012.03.010. Epub 2012 Mar 23. Cell Signal. 2012. PMID: 22465880
-
Regulation of the MIR155 host gene in physiological and pathological processes.Gene. 2013 Dec 10;532(1):1-12. doi: 10.1016/j.gene.2012.12.009. Epub 2012 Dec 14. Gene. 2013. PMID: 23246696 Review.
-
Targeting on glycosylation of mutant FLT3 in acute myeloid leukemia.Hematology. 2019 Dec;24(1):651-660. doi: 10.1080/16078454.2019.1666219. Hematology. 2019. PMID: 31533545 Review.
Cited by
-
Bortezomib inhibits expression of TGF-β1, IL-10, and CXCR4, resulting in decreased survival and migration of cutaneous T cell lymphoma cells.J Immunol. 2015 Mar 15;194(6):2942-53. doi: 10.4049/jimmunol.1402610. Epub 2015 Feb 13. J Immunol. 2015. PMID: 25681335 Free PMC article.
-
Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: from molecular pathogenesis to therapeutic target.Oncotarget. 2015 Mar 20;6(8):5490-500. doi: 10.18632/oncotarget.3545. Oncotarget. 2015. PMID: 25823927 Free PMC article. Review.
-
Interplay between proteasome inhibitors and NF-κB pathway in leukemia and lymphoma: a comprehensive review on challenges ahead of proteasome inhibitors.Cell Commun Signal. 2024 Feb 8;22(1):105. doi: 10.1186/s12964-023-01433-5. Cell Commun Signal. 2024. PMID: 38331801 Free PMC article. Review.
-
NF-κB in Hematological Malignancies.Biomedicines. 2017 May 31;5(2):27. doi: 10.3390/biomedicines5020027. Biomedicines. 2017. PMID: 28561798 Free PMC article. Review.
-
The DAXX co-repressor is directly recruited to active regulatory elements genome-wide to regulate autophagy programs in a model of human prostate cancer.Oncoscience. 2015 Apr 17;2(4):362-72. doi: 10.18632/oncoscience.152. eCollection 2015. Oncoscience. 2015. PMID: 26097870 Free PMC article.
References
-
- Schardt JA, Weber D, Eyholzer M, Mueller BU, Pabst T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin Cancer Res. 2009;15:3834–41. - PubMed
-
- Choudhary C, Olsen JV, Brandts C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36:326–39. - PubMed
-
- Szegezdi E, Macdonald DC, Ní Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
