

Protein intrinsic disorder as a flexible armor and a weapon of HIV-1
- PMID: 22033837
- PMCID: PMC11114566
- DOI: 10.1007/s00018-011-0859-3
Protein intrinsic disorder as a flexible armor and a weapon of HIV-1
Abstract
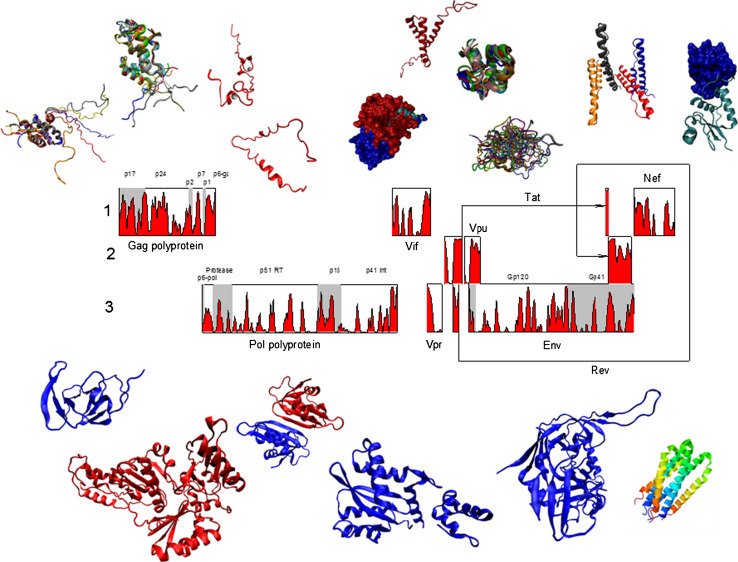
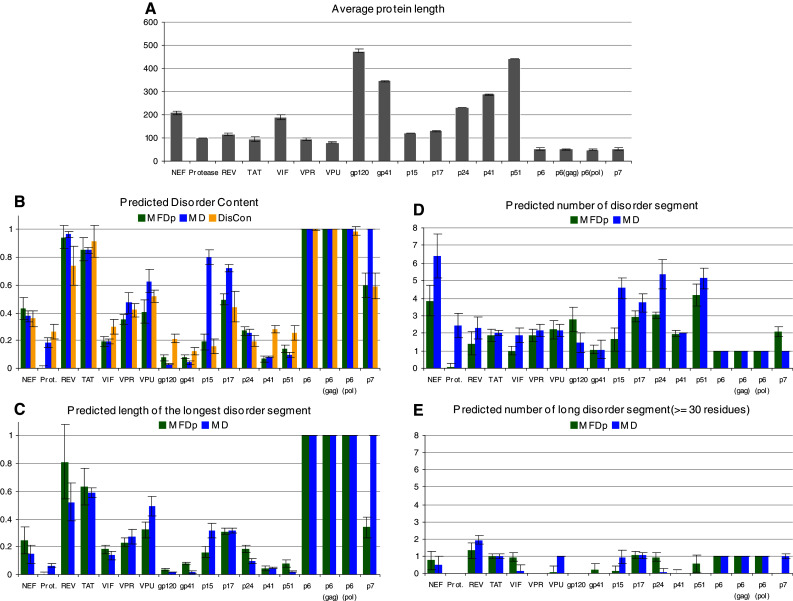
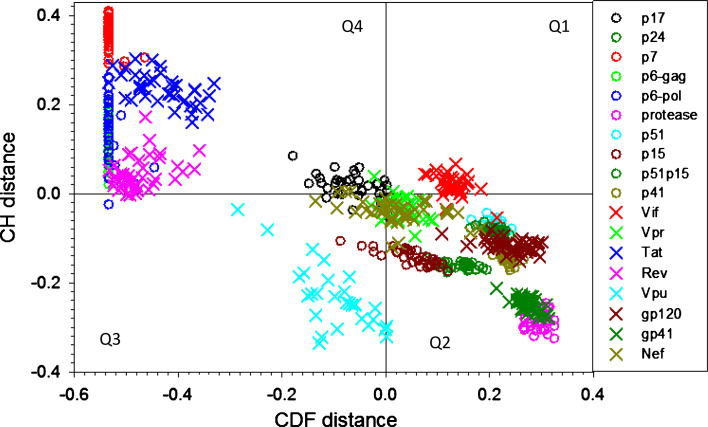
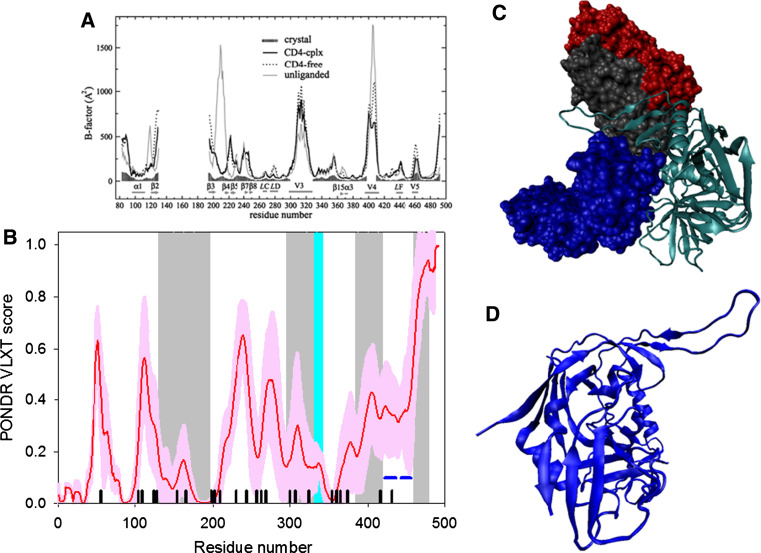
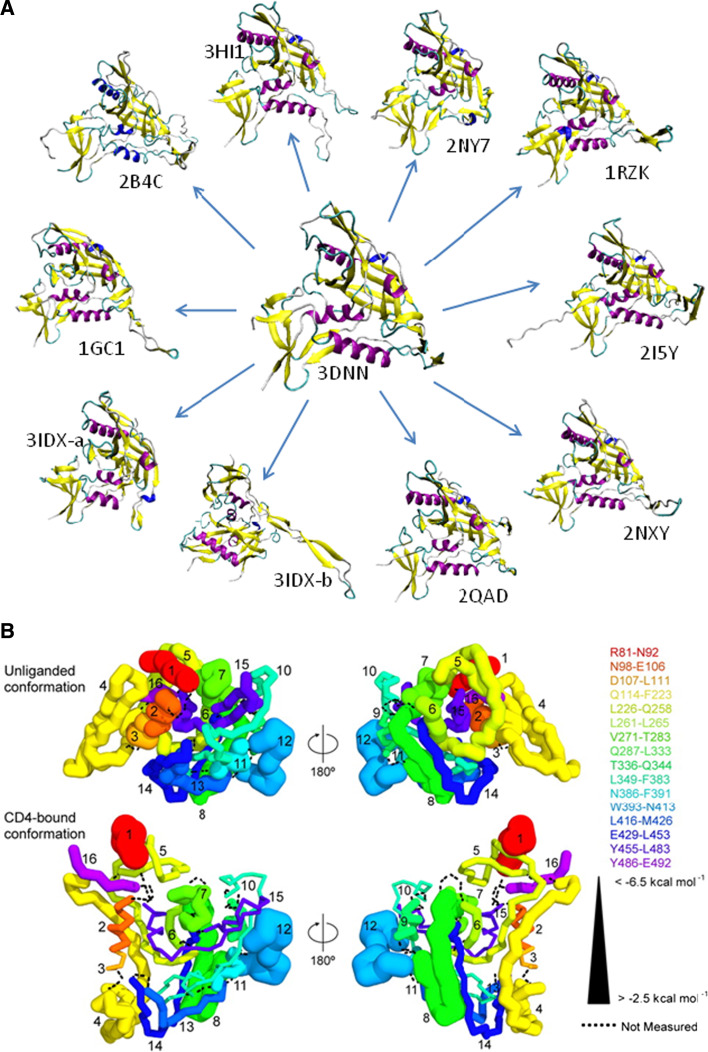
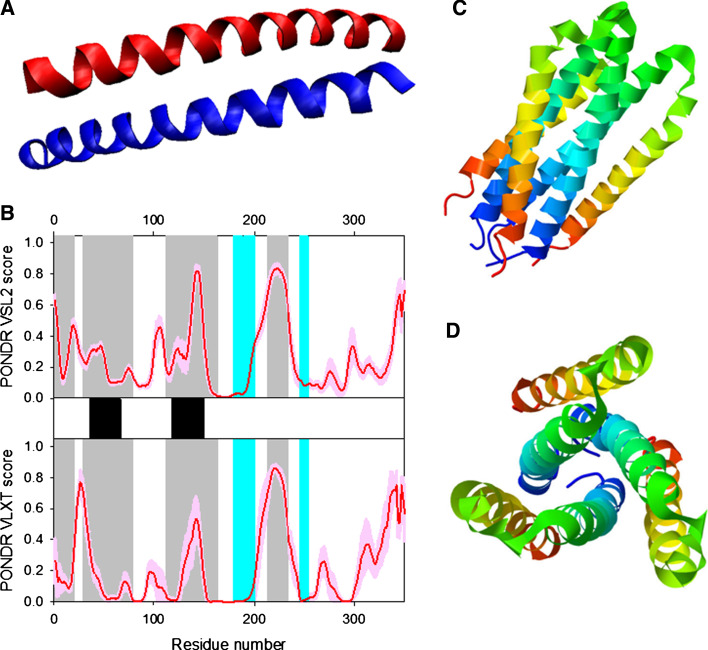
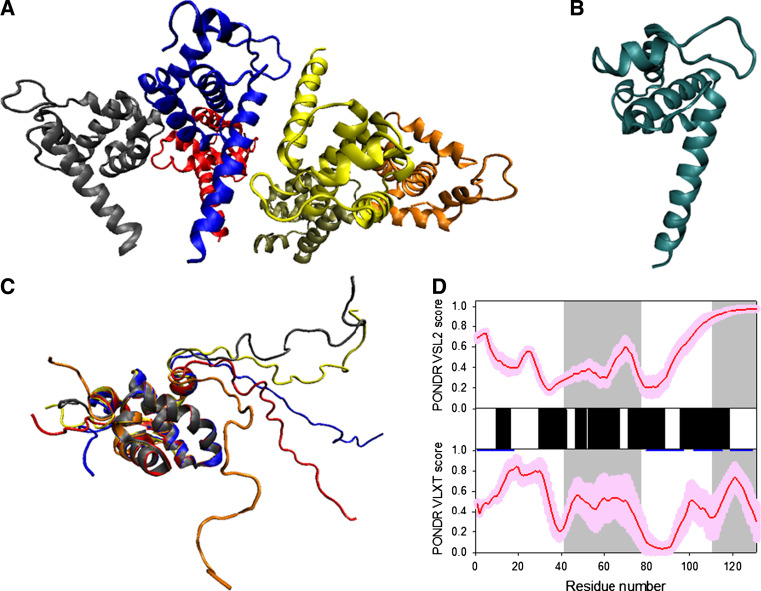
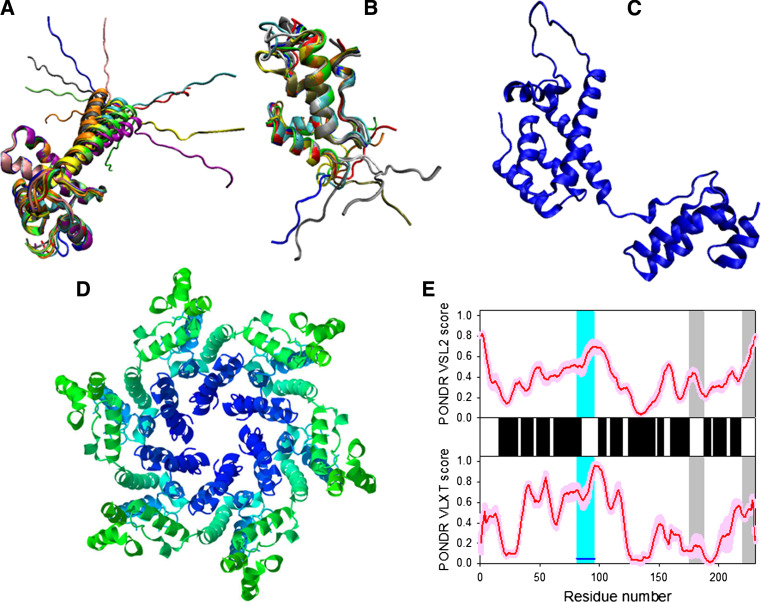
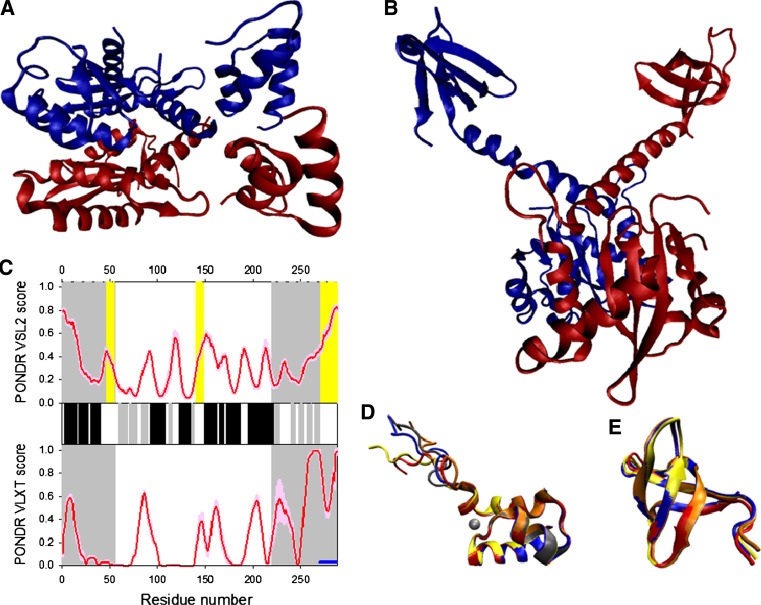
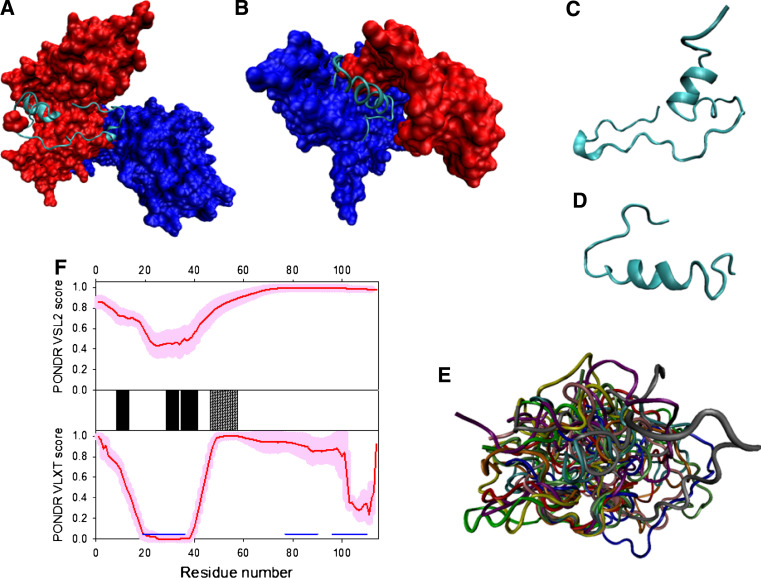
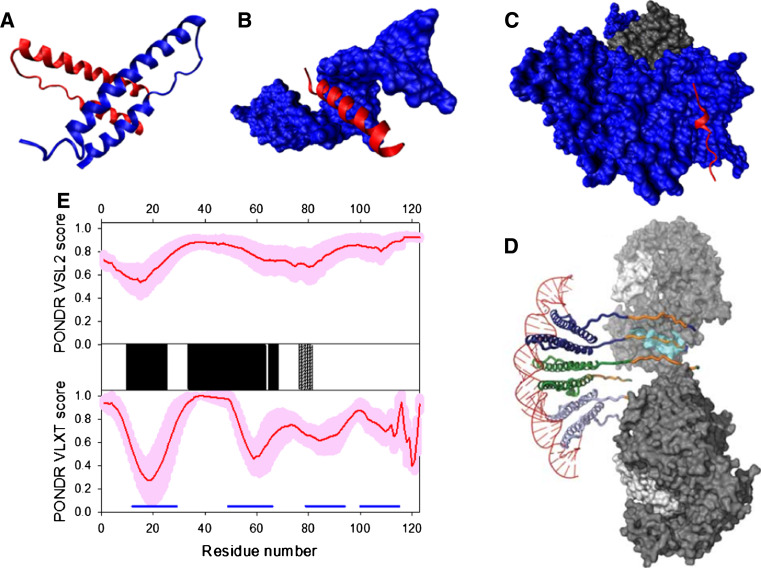
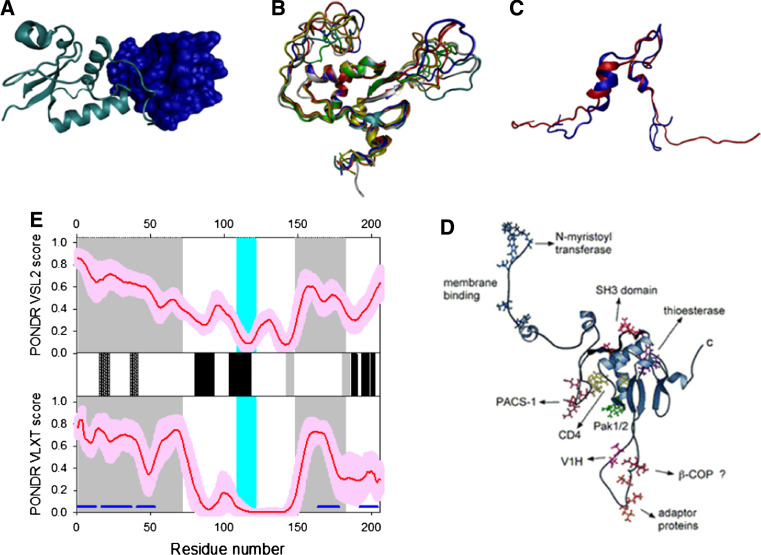
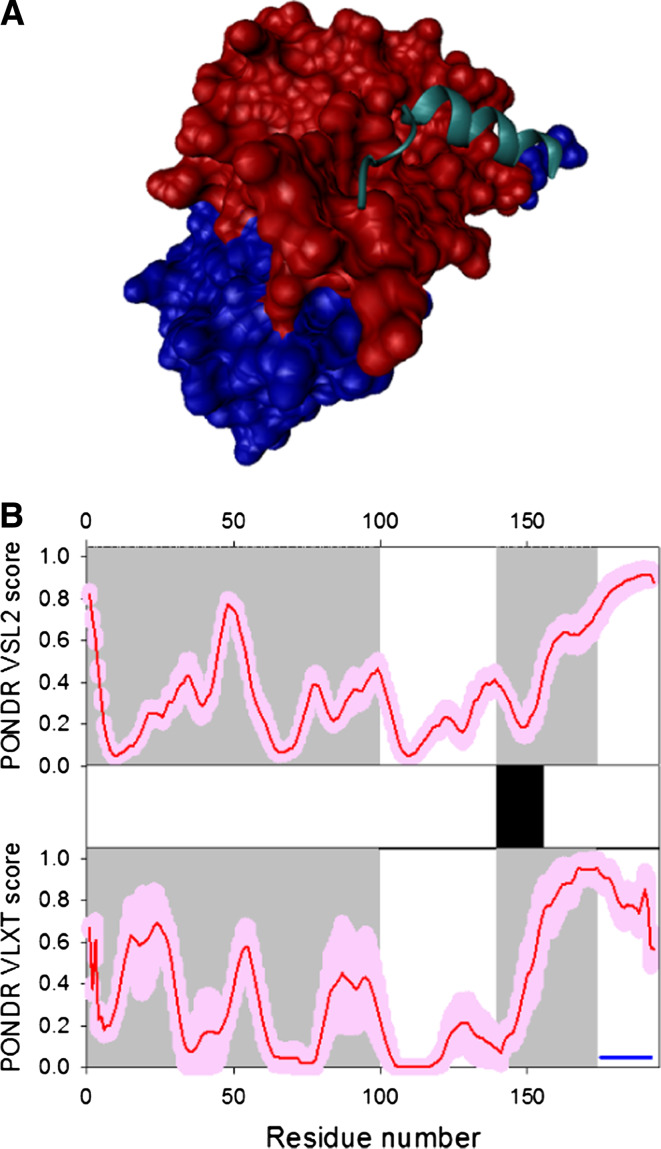
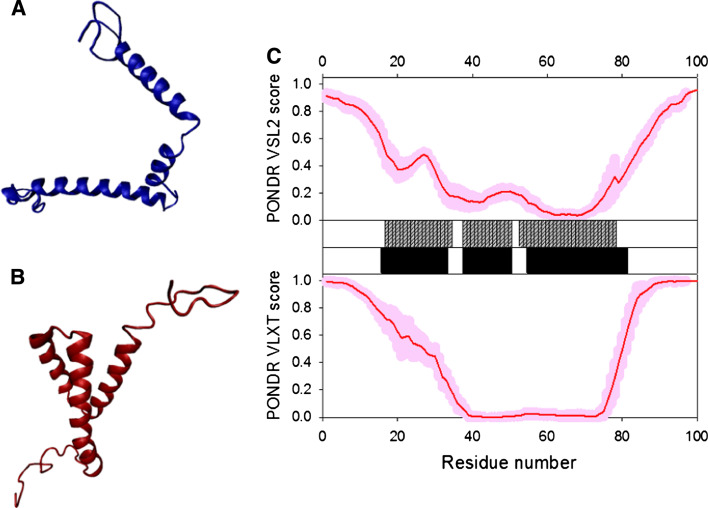
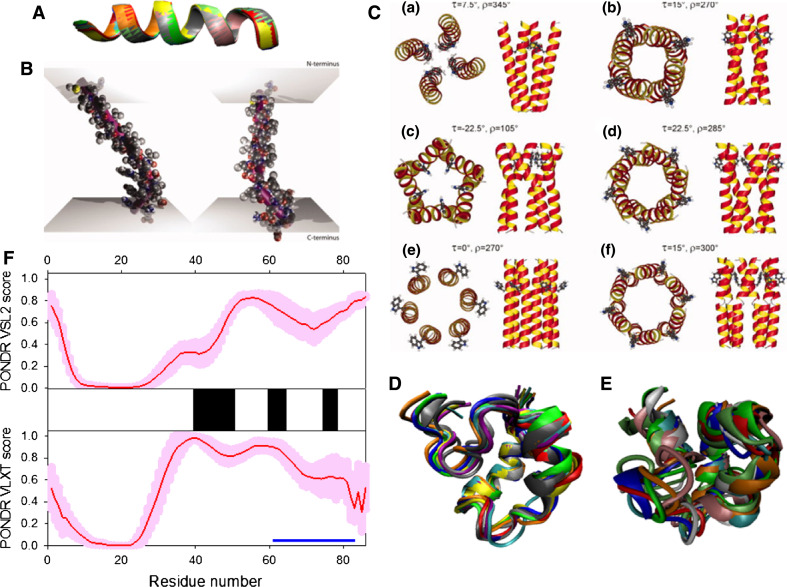
Many proteins and protein regions are disordered in their native, biologically active states. These proteins/regions are abundant in different organisms and carry out important biological functions that complement the functional repertoire of ordered proteins. Viruses, with their highly compact genomes, small proteomes, and high adaptability for fast change in their biological and physical environment utilize many of the advantages of intrinsic disorder. In fact, viral proteins are generally rich in intrinsic disorder, and intrinsically disordered regions are commonly used by viruses to invade the host organisms, to hijack various host systems, and to help viruses in accommodation to their hostile habitats and to manage their economic usage of genetic material. In this review, we focus on the structural peculiarities of HIV-1 proteins, on the abundance of intrinsic disorder in viral proteins, and on the role of intrinsic disorder in their functions.
Figures
Similar articles
-
The intrinsic disorder status of the human hepatitis C virus proteome.Mol Biosyst. 2014 Jun;10(6):1345-63. doi: 10.1039/c4mb00027g. Epub 2014 Apr 22. Mol Biosyst. 2014. PMID: 24752801
-
Viral disorder or disordered viruses: do viral proteins possess unique features?Protein Pept Lett. 2010 Aug;17(8):932-51. doi: 10.2174/092986610791498984. Protein Pept Lett. 2010. PMID: 20450483 Review.
-
Archaic chaos: intrinsically disordered proteins in Archaea.BMC Syst Biol. 2010 May 28;4 Suppl 1(Suppl 1):S1. doi: 10.1186/1752-0509-4-S1-S1. BMC Syst Biol. 2010. PMID: 20522251 Free PMC article.
-
Disordered interactome of human papillomavirus.Curr Pharm Des. 2014;20(8):1274-92. doi: 10.2174/13816128113199990072. Curr Pharm Des. 2014. PMID: 23713779 Review.
-
Role of structural disorder in the multi-functionality of flavivirus proteins.Expert Rev Proteomics. 2022 Mar;19(3):183-196. doi: 10.1080/14789450.2022.2085563. Epub 2022 Jun 7. Expert Rev Proteomics. 2022. PMID: 35655146
Cited by
-
On the Prevalence and Potential Functionality of an Intrinsic Disorder in the MERS-CoV Proteome.Viruses. 2021 Feb 22;13(2):339. doi: 10.3390/v13020339. Viruses. 2021. PMID: 33671602 Free PMC article.
-
DBC1/CCAR2 and CCAR1 Are Largely Disordered Proteins that Have Evolved from One Common Ancestor.Biomed Res Int. 2014;2014:418458. doi: 10.1155/2014/418458. Epub 2014 Dec 11. Biomed Res Int. 2014. PMID: 25610865 Free PMC article.
-
Molecular dynamics simulation in virus research.Front Microbiol. 2012 Jul 19;3:258. doi: 10.3389/fmicb.2012.00258. eCollection 2012. Front Microbiol. 2012. PMID: 22833741 Free PMC article.
-
DISOselect: Disorder predictor selection at the protein level.Protein Sci. 2020 Jan;29(1):184-200. doi: 10.1002/pro.3756. Epub 2019 Nov 7. Protein Sci. 2020. PMID: 31642118 Free PMC article.
-
Computational Prediction of Intrinsic Disorder in Protein Sequences with the disCoP Meta-predictor.Methods Mol Biol. 2020;2141:21-35. doi: 10.1007/978-1-0716-0524-0_2. Methods Mol Biol. 2020. PMID: 32696351
References
-
- Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998;3:473–484. - PubMed
-
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/S1093-3263(00)00138-8. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
