

Inflammasomes in intestinal inflammation and cancer
- PMID: 22005480
- PMCID: PMC3442608
- DOI: 10.1053/j.gastro.2011.10.002
Inflammasomes in intestinal inflammation and cancer
Abstract
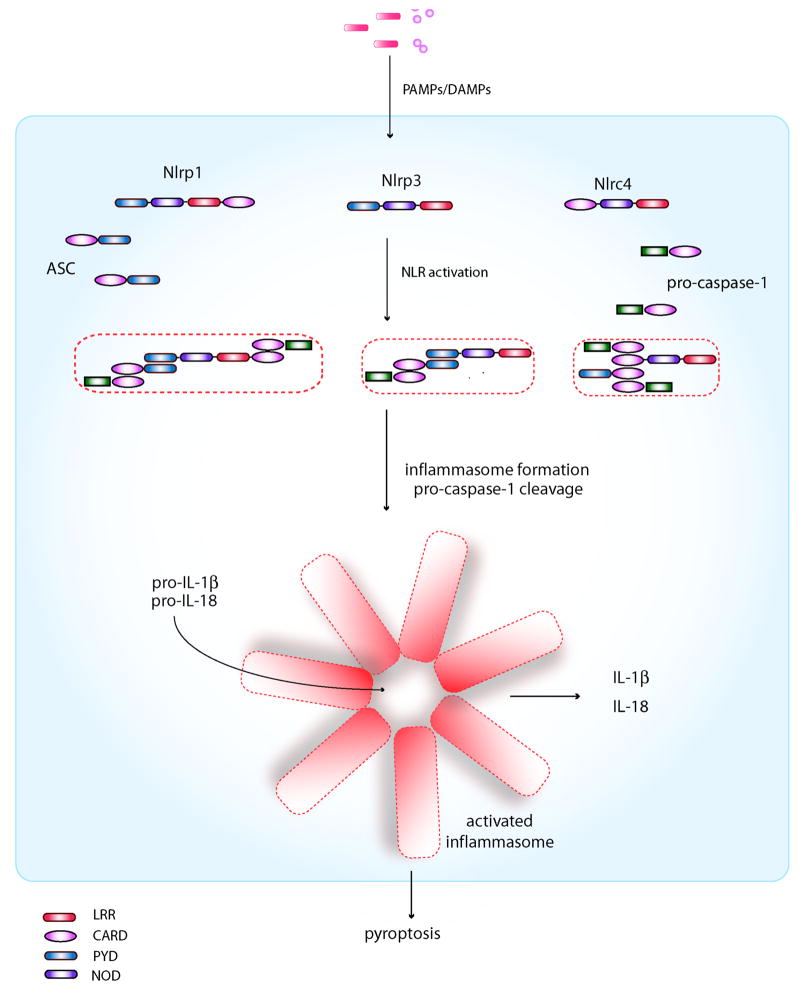
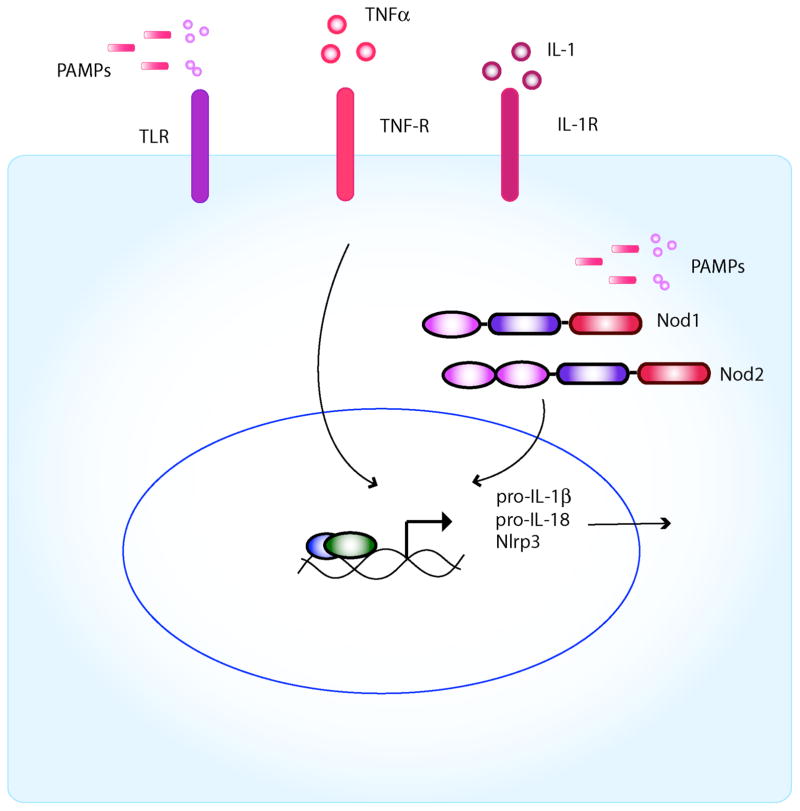
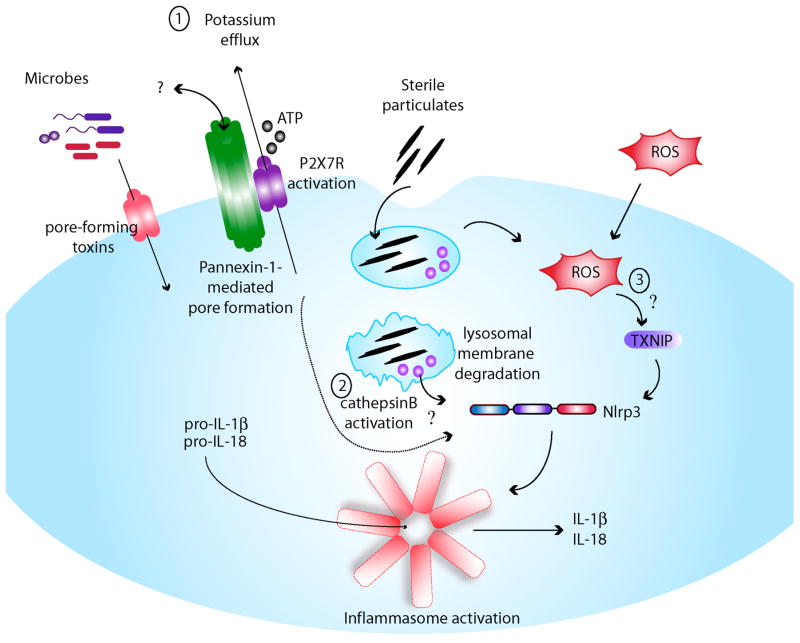
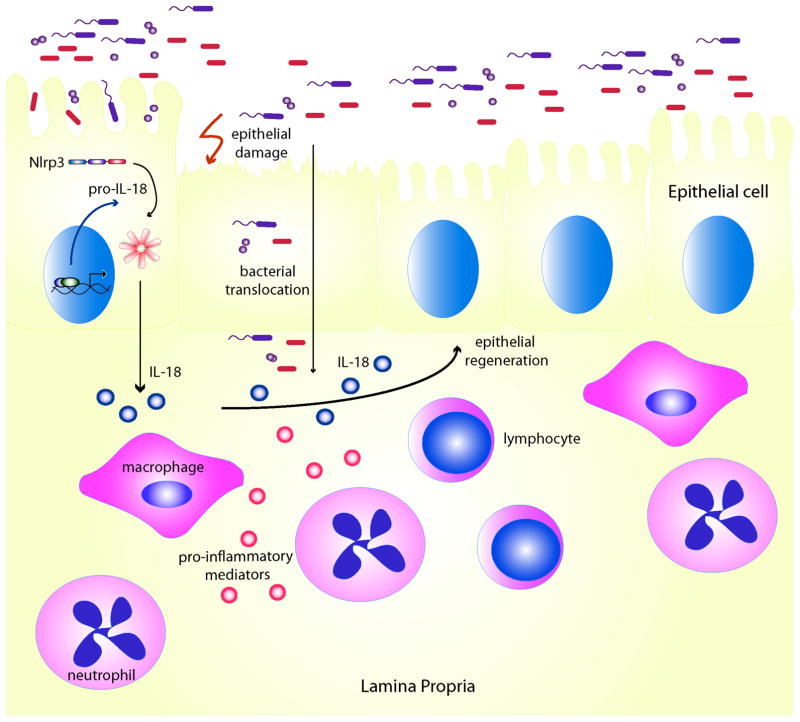
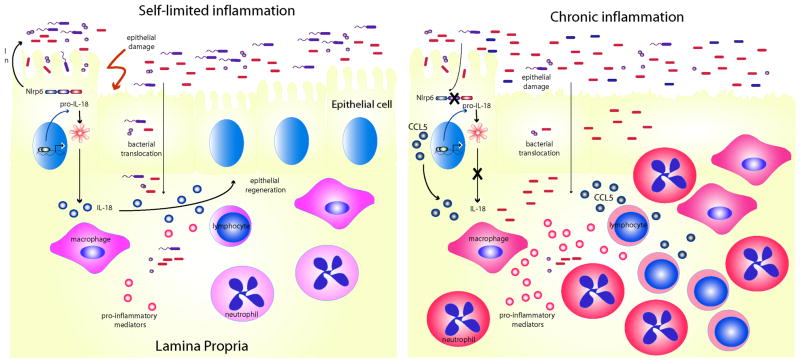
Inflammasomes are multi-protein complexes that mediate activation of caspase-1, which promotes secretion of the proinflammatory cytokines interleukin-1β and interleukin-18 and pyroptosis, a form of phagocyte cell death induced by bacterial pathogens. Members of the Nod-like receptor family (including Nlrp1, Nlrp3, and Nlrc4), the DNA sensor Aim2, the adaptor apoptosis-associated speck-like protein (ASC), and pro-caspase-1 are important components of inflammasomes. Stimulation with specific microbial and endogenous molecules leads to inflammasome assembly and caspase-1 activation. Inflammasomes are believed to mediate host defense against microbial pathogens and tissue homeostasis within the intestine, and their dysregulation might contribute to inflammatory diseases and intestinal cancer. Improving our understanding of inflammasome signaling pathways could provide insights into the pathogenesis of many gastrointestinal disorders and the development of therapeutic targets and approaches to treat diseases such as inflammatory bowel diseases and gastrointestinal cancers.
Copyright © 2011 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
[Inflammatory bowel diseases and inflammasome].Korean J Gastroenterol. 2011 Dec;58(6):300-10. doi: 10.4166/kjg.2011.58.6.300. Korean J Gastroenterol. 2011. PMID: 22198227 Review. Korean.
-
Cytokine Secretion and Pyroptosis of Thyroid Follicular Cells Mediated by Enhanced NLRP3, NLRP1, NLRC4, and AIM2 Inflammasomes Are Associated With Autoimmune Thyroiditis.Front Immunol. 2018 Jun 4;9:1197. doi: 10.3389/fimmu.2018.01197. eCollection 2018. Front Immunol. 2018. PMID: 29915579 Free PMC article.
-
Inflammasomes and Fibrosis.Front Immunol. 2021 Jun 11;12:643149. doi: 10.3389/fimmu.2021.643149. eCollection 2021. Front Immunol. 2021. PMID: 34177893 Free PMC article. Review.
-
Sensing and reacting to microbes through the inflammasomes.Nat Immunol. 2012 Mar 19;13(4):325-32. doi: 10.1038/ni.2231. Nat Immunol. 2012. PMID: 22430785 Free PMC article. Review.
-
Altered expression of inflammasomes in Hirschsprung's disease.Pediatr Surg Int. 2019 Jan;35(1):15-20. doi: 10.1007/s00383-018-4371-9. Epub 2018 Nov 1. Pediatr Surg Int. 2019. PMID: 30386901
Cited by
-
Inflammation and Cell Death During Cholestasis: The Evolving Role of Bile Acids.Gene Expr. 2019 Nov 4;19(3):215-228. doi: 10.3727/105221619X15614873062730. Epub 2019 Jun 28. Gene Expr. 2019. PMID: 31253204 Free PMC article. Review.
-
NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation.Sci Rep. 2020 Jan 13;10(1):198. doi: 10.1038/s41598-019-57043-0. Sci Rep. 2020. PMID: 31932628 Free PMC article.
-
A Potential Role of Salmonella Infection in the Onset of Inflammatory Bowel Diseases.Front Immunol. 2017 Feb 28;8:191. doi: 10.3389/fimmu.2017.00191. eCollection 2017. Front Immunol. 2017. PMID: 28293241 Free PMC article. Review.
-
Inflammatory caspase-related pyroptosis: mechanism, regulation and therapeutic potential for inflammatory bowel disease.Gastroenterol Rep (Oxf). 2018 Aug;6(3):167-176. doi: 10.1093/gastro/goy011. Epub 2018 May 2. Gastroenterol Rep (Oxf). 2018. PMID: 30151200 Free PMC article. Review.
-
Inflammation, Autoinflammation and Autoimmunity in Inflammatory Bowel Diseases.Curr Issues Mol Biol. 2023 Jun 30;45(7):5534-5557. doi: 10.3390/cimb45070350. Curr Issues Mol Biol. 2023. PMID: 37504266 Free PMC article. Review.
References
-
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. - PubMed
-
- Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–10. - PubMed
-
- Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA46592-22S3/CA/NCI NIH HHS/United States
- CA133185/CA/NCI NIH HHS/United States
- P30 CA046592/CA/NCI NIH HHS/United States
- R01 DK091191/DK/NIDDK NIH HHS/United States
- AR051790/AR/NIAMS NIH HHS/United States
- R01 DK061707/DK/NIDDK NIH HHS/United States
- DK091191/DK/NIDDK NIH HHS/United States
- AR059688/AR/NIAMS NIH HHS/United States
- AI06331/AI/NIAID NIH HHS/United States
- R01 AR059688/AR/NIAMS NIH HHS/United States
- K08 CA133185/CA/NCI NIH HHS/United States
- R56 AI063331/AI/NIAID NIH HHS/United States
- DK61707/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
