

Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes
- PMID: 21944046
- PMCID: PMC3188835
- DOI: 10.1016/j.ajhg.2011.08.011
Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes
Abstract
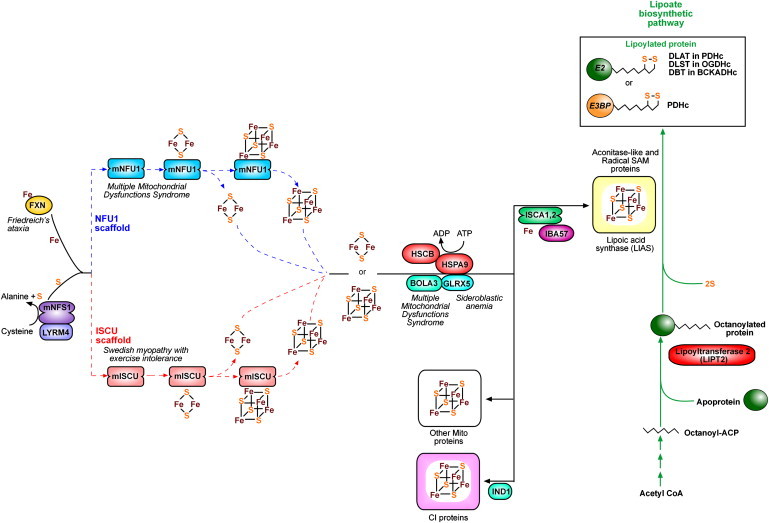
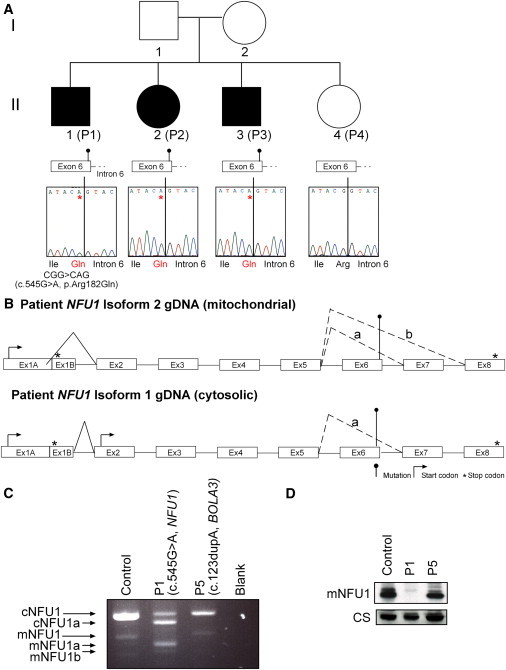
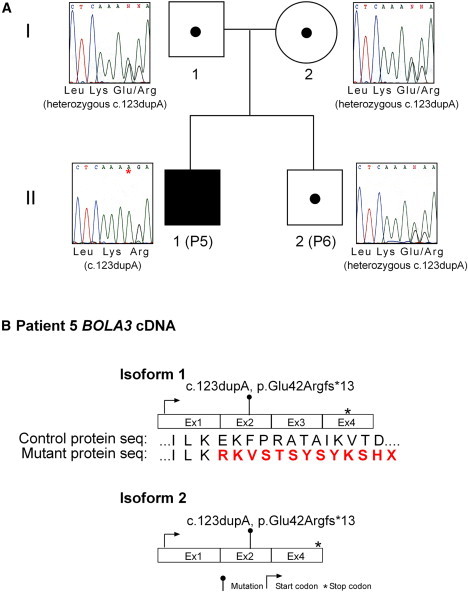
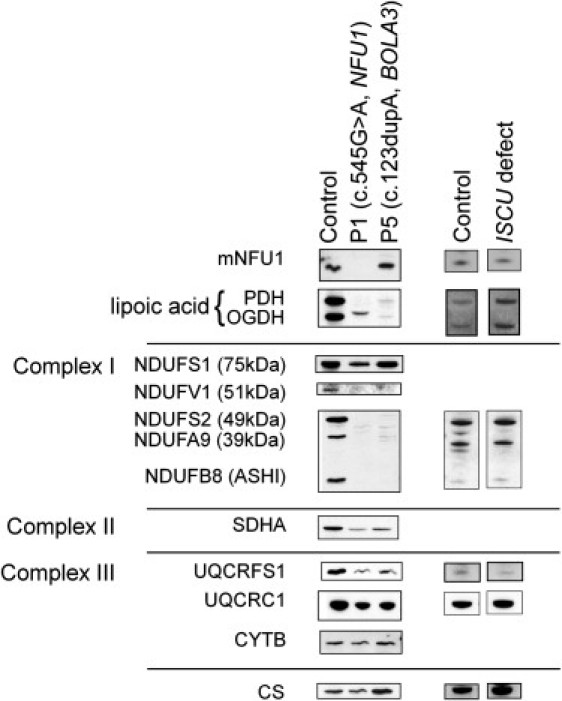
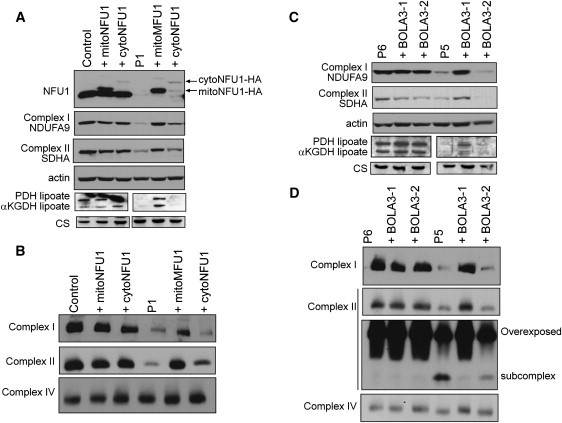
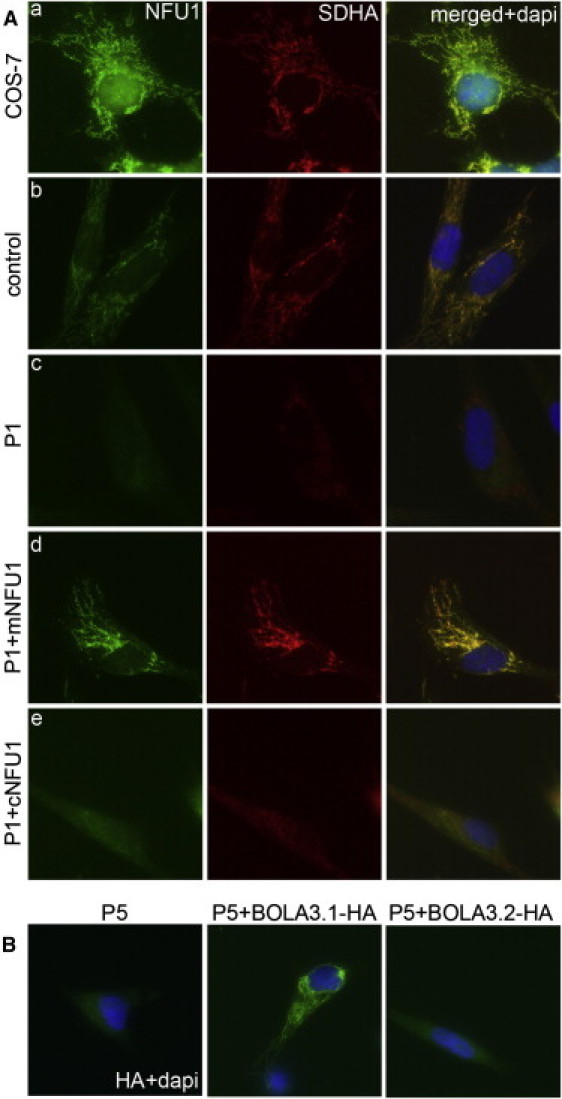
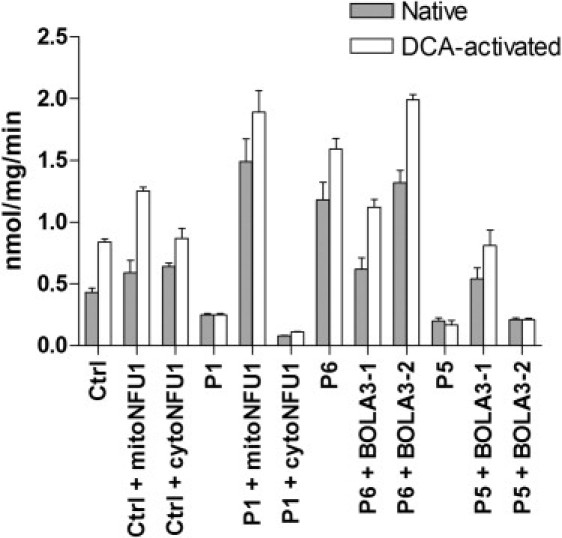
Severe combined deficiency of the 2-oxoacid dehydrogenases, associated with a defect in lipoate synthesis and accompanied by defects in complexes I, II, and III of the mitochondrial respiratory chain, is a rare autosomal recessive syndrome with no obvious causative gene defect. A candidate locus for this syndrome was mapped to chromosomal region 2p14 by microcell-mediated chromosome transfer in two unrelated families. Unexpectedly, analysis of genes in this area identified mutations in two different genes, both of which are involved in [Fe-S] cluster biogenesis. A homozygous missense mutation, c.545G>A, near the splice donor of exon 6 in NFU1 predicting a p.Arg182Gln substitution was found in one of the families. The mutation results in abnormal mRNA splicing of exon 6, and no mature protein could be detected in fibroblast mitochondria. A single base-pair duplication c.123dupA was identified in BOLA3 in the second family, causing a frame shift that produces a premature stop codon (p.Glu42Argfs(∗)13). Transduction of fibroblast lines with retroviral vectors expressing the mitochondrial, but not the cytosolic isoform of NFU1 and with isoform 1, but not isoform 2 of BOLA3 restored both respiratory chain function and oxoacid dehydrogenase complexes. NFU1 was previously proposed to be an alternative scaffold to ISCU for the biogenesis of [Fe-S] centers in mitochondria, and the function of BOLA3 was previously unknown. Our results demonstrate that both play essential roles in the production of [Fe-S] centers for the normal maturation of lipoate-containing 2-oxoacid dehydrogenases, and for the assembly of the respiratory chain complexes.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Impact of mutations within the [Fe-S] cluster or the lipoic acid biosynthesis pathways on mitochondrial protein expression profiles in fibroblasts from patients.Mol Genet Metab. 2017 Nov;122(3):85-94. doi: 10.1016/j.ymgme.2017.08.001. Epub 2017 Aug 3. Mol Genet Metab. 2017. PMID: 28803783
-
A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins.Am J Hum Genet. 2011 Nov 11;89(5):656-67. doi: 10.1016/j.ajhg.2011.10.005. Am J Hum Genet. 2011. PMID: 22077971 Free PMC article.
-
Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings.J Inherit Metab Dis. 2013 Jan;36(1):55-62. doi: 10.1007/s10545-012-9489-7. Epub 2012 May 5. J Inherit Metab Dis. 2013. PMID: 22562699
-
Lipoic acid biosynthesis defects.J Inherit Metab Dis. 2014 Jul;37(4):553-63. doi: 10.1007/s10545-014-9705-8. Epub 2014 Apr 29. J Inherit Metab Dis. 2014. PMID: 24777537 Review.
-
Mitochondrial iron-sulfur protein biogenesis and human disease.Biochimie. 2014 May;100:61-77. doi: 10.1016/j.biochi.2014.01.010. Epub 2014 Jan 23. Biochimie. 2014. PMID: 24462711 Review.
Cited by
-
Dissecting a complex chemical stress: chemogenomic profiling of plant hydrolysates.Mol Syst Biol. 2013 Jun 18;9:674. doi: 10.1038/msb.2013.30. Mol Syst Biol. 2013. PMID: 23774757 Free PMC article.
-
Novel mutations in IBA57 are associated with leukodystrophy and variable clinical phenotypes.J Neurol. 2017 Jan;264(1):102-111. doi: 10.1007/s00415-016-8312-z. Epub 2016 Oct 26. J Neurol. 2017. PMID: 27785568
-
Pitfalls of relying on genetic testing only to diagnose inherited metabolic disorders in non-western populations - 5 cases of pyruvate dehydrogenase deficiency from South Africa.Mol Genet Metab Rep. 2020 Jul 22;24:100629. doi: 10.1016/j.ymgmr.2020.100629. eCollection 2020 Sep. Mol Genet Metab Rep. 2020. PMID: 32742935 Free PMC article.
-
Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes.Hum Mol Genet. 2013 Nov 15;22(22):4460-73. doi: 10.1093/hmg/ddt295. Epub 2013 Jun 28. Hum Mol Genet. 2013. PMID: 23814038 Free PMC article.
-
Genetic screens reveal a central role for heme metabolism in artemisinin susceptibility.Nat Commun. 2020 Sep 23;11(1):4813. doi: 10.1038/s41467-020-18624-0. Nat Commun. 2020. PMID: 32968076 Free PMC article.
References
-
- Cameron J.M., Levandovskiy V., Mackay N., Tein I., Robinson B.H. Deficiency of pyruvate dehydrogenase caused by novel and known mutations in the E1alpha subunit. Am. J. Med. Genet. A. 2004;131:59–66. - PubMed
-
- Lissens W., De Meirleir L., Seneca S., Liebaers I., Brown G.K., Brown R.M., Ito M., Naito E., Kuroda Y., Kerr D.S. Mutations in the X-linked pyruvate dehydrogenase (E1) alpha subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum. Mutat. 2000;15:209–219. - PubMed
-
- Kerr D.S., Wexler I.D., Tripatara A., Patel M.S. Human defects of the pyruvate dehydrogenase complex. In: Patel M.S., Harris R.A., editors. Alpha-keto acid dehydrogenase complexes, R.T. Birkhäuser; Basel: 1996. pp. 249–269.
-
- Maj M.C., MacKay N., Levandovskiy V., Addis J., Baumgartner E.R., Baumgartner M.R., Robinson B.H., Cameron J.M. Pyruvate dehydrogenase phosphatase deficiency: Identification of the first mutation in two brothers and restoration of activity by protein complementation. J. Clin. Endocrinol. Metab. 2005;90:4101–4107. - PubMed
-
- Robinson B.H. Lactic acidemia (Disorders of pyruvate carboxylase, pyruvate dehydrogenase) In: Scriver C.R., Sly W.S., Valle D., editors. The metabolic and molecular bases of inherited disease, B.A. McGraw-Hill; New York: 2001. pp. 2275–2295.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
