

Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling
- PMID: 21937694
- PMCID: PMC3265205
- DOI: 10.1182/blood-2011-06-357384
Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling
Abstract
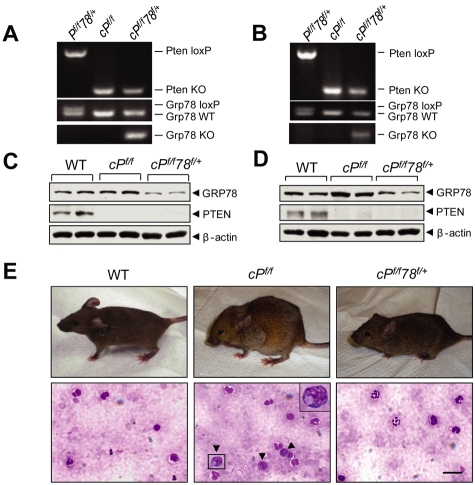
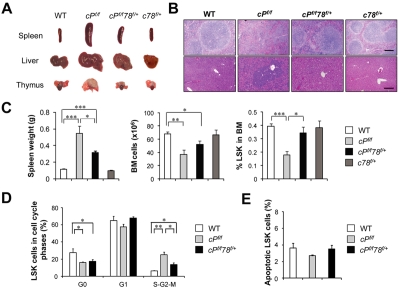
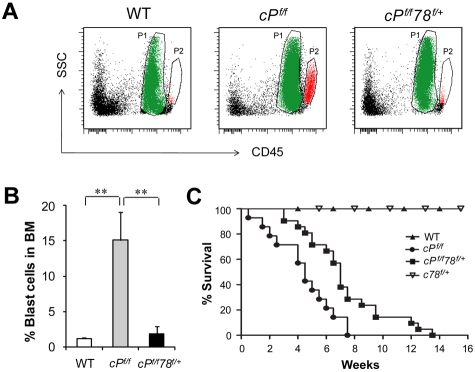
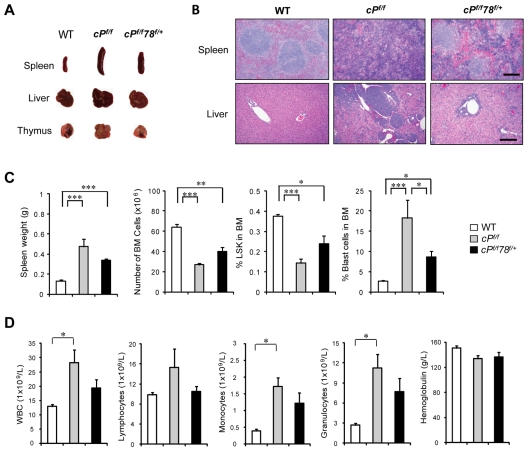
Traditionally, GRP78 is regarded as protective against hypoxia and nutrient starvation prevalent in the microenvironment of solid tumors; thus, its role in the development of hematologic malignancies remains to be determined. To directly elucidate the requirement of GRP78 in leukemogenesis, we created a biallelic conditional knockout mouse model of GRP78 and PTEN in the hematopoietic system. Strikingly, heterozygous knockdown of GRP78 in PTEN null mice is sufficient to restore the hematopoietic stem cell population back to the normal percentage and suppress leukemic blast cell expansion. AKT/mTOR activation in PTEN null BM cells is potently inhibited by Grp78 heterozygosity, corresponding with suppression of the PI3K/AKT pathway by GRP78 knockdown in leukemia cell lines. This is the first demonstration that GRP78 is a critical effector of leukemia progression, at least in part through regulation of oncogenic PI3K/AKT signaling. In agreement with PI3K/AKT as an effector for cytosine arabinoside resistance in acute myeloid leukemia, overexpression of GRP78 renders human leukemic cells more resistant to cytosine arabinoside-induced apoptosis, whereas knockdown of GRP78 sensitizes them. These, coupled with the emerging association of elevated GRP78 expression in leukemic blasts of adult patients and early relapse in childhood leukemia, suggest that GRP78 is a novel therapeutic target for leukemia.
Figures
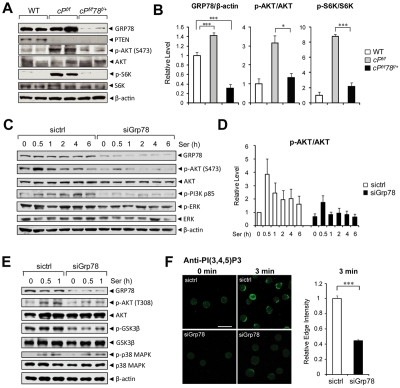
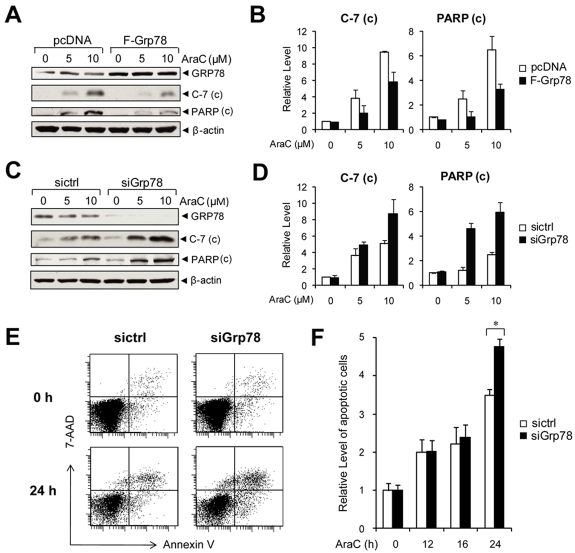
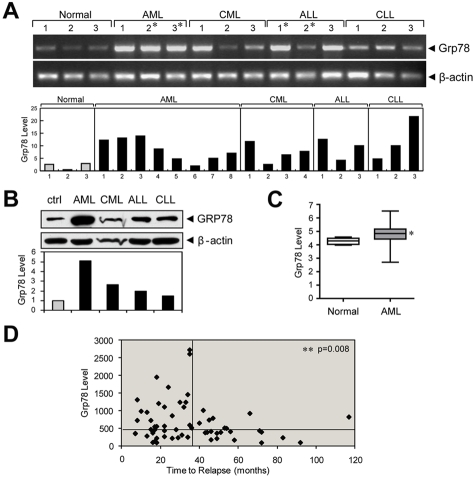
Comment in
-
GRPling with PTEN.Blood. 2012 Jan 19;119(3):648-9. doi: 10.1182/blood-2011-11-382614. Blood. 2012. PMID: 22262740 No abstract available.
Similar articles
-
Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium.Proc Natl Acad Sci U S A. 2008 Dec 9;105(49):19444-9. doi: 10.1073/pnas.0807691105. Epub 2008 Nov 25. Proc Natl Acad Sci U S A. 2008. PMID: 19033462 Free PMC article.
-
GRPling with PTEN.Blood. 2012 Jan 19;119(3):648-9. doi: 10.1182/blood-2011-11-382614. Blood. 2012. PMID: 22262740 No abstract available.
-
Activation of the PI3K/AKT pathway induces urothelial carcinoma of the renal pelvis: identification in human tumors and confirmation in animal models.Cancer Res. 2009 Nov 1;69(21):8256-64. doi: 10.1158/0008-5472.CAN-09-1689. Epub 2009 Oct 20. Cancer Res. 2009. PMID: 19843858 Free PMC article.
-
Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway.Leukemia. 2011 Jul;25(7):1064-79. doi: 10.1038/leu.2011.46. Epub 2011 Mar 25. Leukemia. 2011. PMID: 21436840 Review.
-
PI3K Targeting in Non-solid Cancer.Curr Top Microbiol Immunol. 2022;436:393-407. doi: 10.1007/978-3-031-06566-8_17. Curr Top Microbiol Immunol. 2022. PMID: 36243854 Free PMC article. Review.
Cited by
-
ER Stress Response and Induction of Apoptosis in Malignant Pleural Mesothelioma: The Achilles Heel Targeted by the Anticancer Ruthenium Drug BOLD-100.Cancers (Basel). 2022 Aug 26;14(17):4126. doi: 10.3390/cancers14174126. Cancers (Basel). 2022. PMID: 36077664 Free PMC article.
-
The Unfolded Protein Response: A Novel Therapeutic Target in Acute Leukemias.Cancers (Basel). 2020 Feb 1;12(2):333. doi: 10.3390/cancers12020333. Cancers (Basel). 2020. PMID: 32024211 Free PMC article. Review.
-
Modelling the effect of GRP78 on anti-oestrogen sensitivity and resistance in breast cancer.Interface Focus. 2013 Aug 6;3(4):20130012. doi: 10.1098/rsfs.2013.0012. Interface Focus. 2013. PMID: 24511377 Free PMC article.
-
Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis.J Cell Physiol. 2015 Jul;230(7):1413-20. doi: 10.1002/jcp.24923. J Cell Physiol. 2015. PMID: 25546813 Free PMC article. Review.
-
AKT inhibition mitigates GRP78 (glucose-regulated protein) expression and contribution to chemoresistance in endometrial cancers.Int J Cancer. 2013 Jul;133(1):21-30. doi: 10.1002/ijc.27994. Epub 2013 Feb 8. Int J Cancer. 2013. PMID: 23280503 Free PMC article.
References
-
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–13378. - PubMed
-
- Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22(14):2954–2963. - PubMed
-
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133(3):403–414. - PubMed
-
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. - PubMed
-
- Lesche R, Groszer M, Gao J, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32(2):148–149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
