

Functional and physical interaction between the mismatch repair and FA-BRCA pathways
- PMID: 21865299
- PMCID: PMC3196888
- DOI: 10.1093/hmg/ddr366
Functional and physical interaction between the mismatch repair and FA-BRCA pathways
Abstract
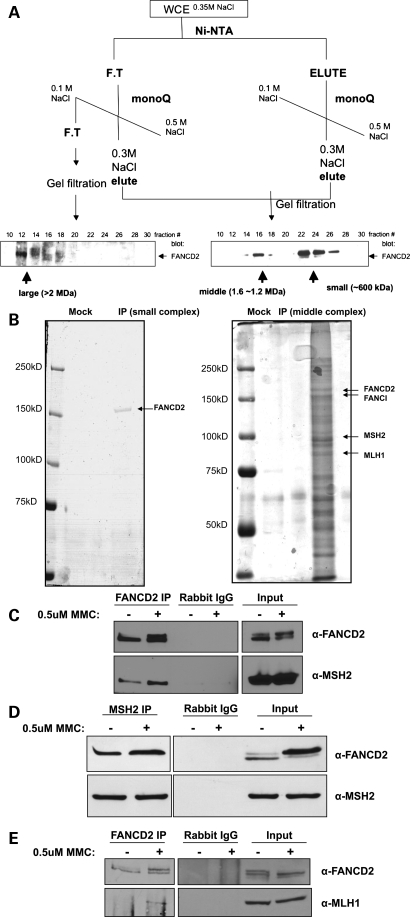
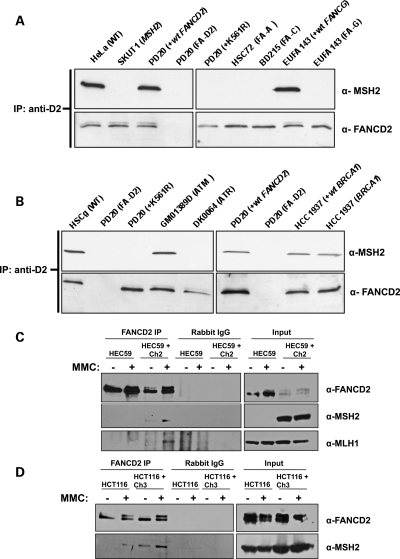
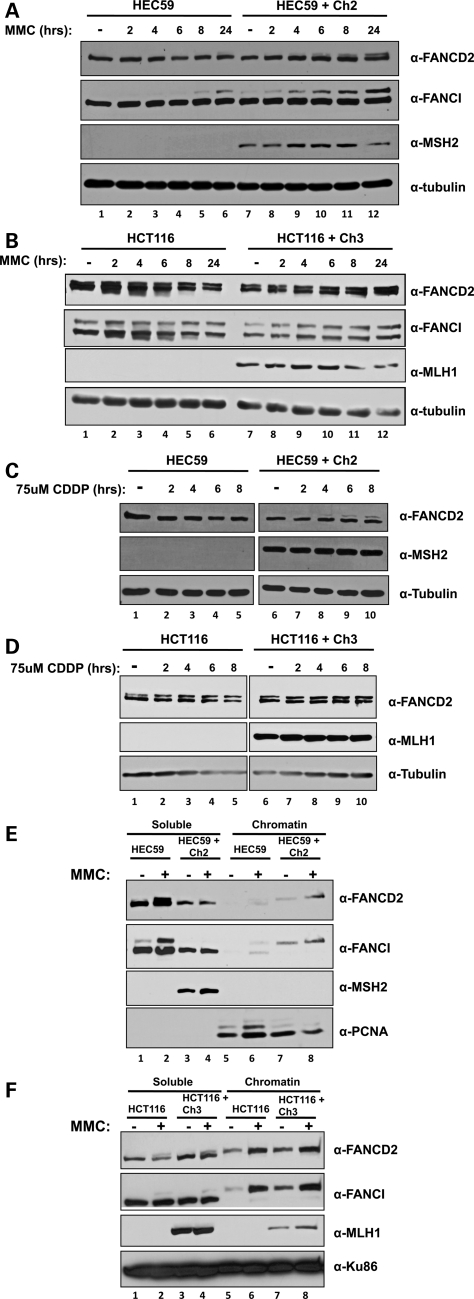
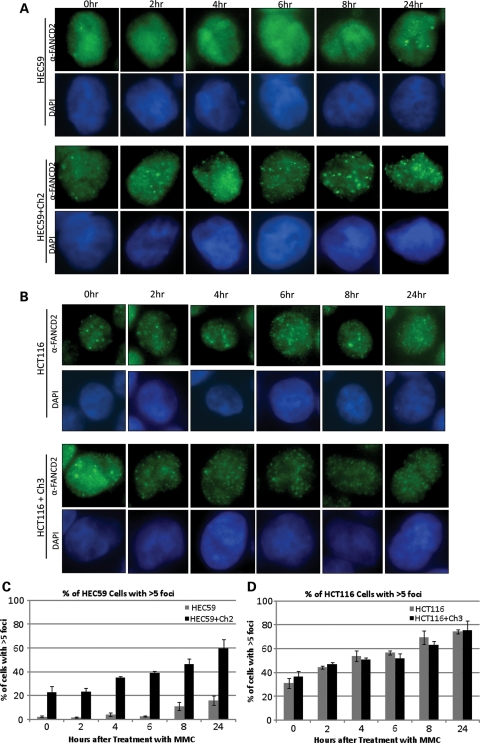
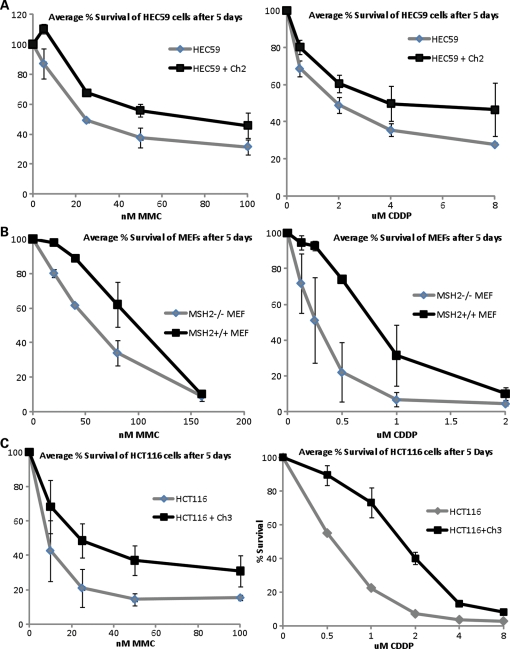
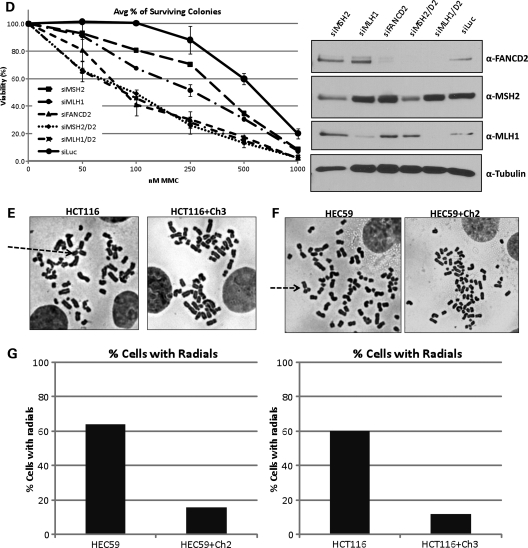
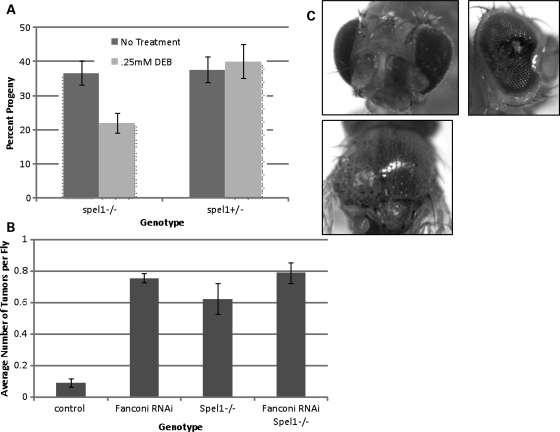
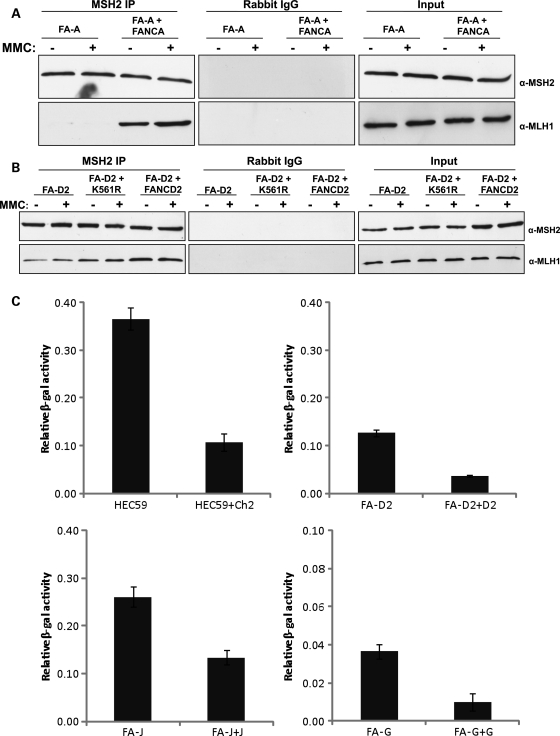
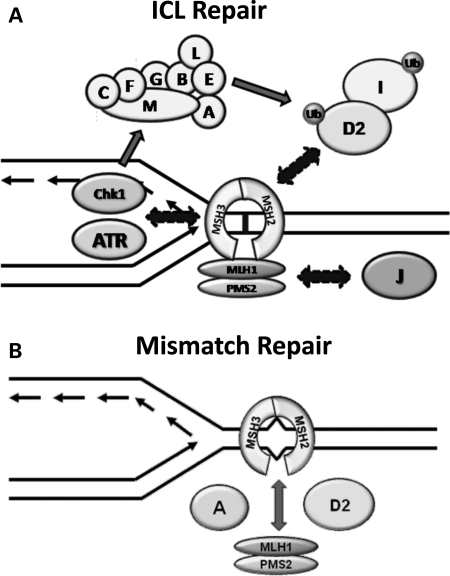
Fanconi anemia (FA) is a rare genetic disorder characterized by bone marrow failure and an increased risk for leukemia and cancer. Fifteen proteins thought to function in the repair of DNA interstrand crosslinks (ICLs) comprise what is known as the FA-BRCA pathway. Activation of this pathway leads to the monoubiquitylation and chromatin localization of FANCD2 and FANCI. It has previously been shown that FANCJ interacts with the mismatch repair (MMR) complex MutLα. Here we show that FANCD2 interacts with the MMR proteins MSH2 and MLH1. FANCD2 monoubiquitylation, foci formation and chromatin loading are greatly diminished in MSH2-deficient cells. Human or mouse cells lacking MSH2 or MLH1 display increased sensitivity and radial formation in response to treatment with DNA crosslinking agents. Studies in human cell lines and Drosophila mutants suggest an epistatic relationship between FANCD2, MSH2 and MLH1 with regard to ICL repair. Surprisingly, the interaction between MSH2 and MLH1 is compromised in multiple FA cell lines, and FA cell lines exhibit deficient MMR. These results suggest a significant role for MMR proteins in the activation of the FA pathway and repair of ICLs. In addition, we provide the first evidence for a defect in MMR in FA cell lines.
Figures
Similar articles
-
What is wrong with Fanconi anemia cells?Cell Cycle. 2014;13(24):3823-7. doi: 10.4161/15384101.2014.980633. Cell Cycle. 2014. PMID: 25486020 Free PMC article. Review.
-
Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses.EMBO J. 2014 Aug 1;33(15):1698-712. doi: 10.15252/embj.201387530. Epub 2014 Jun 25. EMBO J. 2014. PMID: 24966277 Free PMC article.
-
The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells.EMBO J. 2007 Jul 11;26(13):3238-49. doi: 10.1038/sj.emboj.7601754. Epub 2007 Jun 21. EMBO J. 2007. PMID: 17581638 Free PMC article.
-
Differential operation of MLH1/MSH2 and FANCD2 crosstalk in chemotolerant bladder carcinoma: a clinical and therapeutic intervening study.Mol Cell Biochem. 2023 Jul;478(7):1599-1610. doi: 10.1007/s11010-022-04616-9. Epub 2022 Nov 24. Mol Cell Biochem. 2023. Retraction in: Mol Cell Biochem. 2024 Jul;479(7):1865. doi: 10.1007/s11010-024-05021-0 PMID: 36434146 Retracted.
-
Ubiquitylation and the Fanconi anemia pathway.FEBS Lett. 2011 Sep 16;585(18):2853-60. doi: 10.1016/j.febslet.2011.04.078. Epub 2011 May 19. FEBS Lett. 2011. PMID: 21605559 Free PMC article. Review.
Cited by
-
What is wrong with Fanconi anemia cells?Cell Cycle. 2014;13(24):3823-7. doi: 10.4161/15384101.2014.980633. Cell Cycle. 2014. PMID: 25486020 Free PMC article. Review.
-
FAN1 controls mismatch repair complex assembly via MLH1 retention to stabilize CAG repeat expansion in Huntington's disease.Cell Rep. 2021 Aug 31;36(9):109649. doi: 10.1016/j.celrep.2021.109649. Cell Rep. 2021. PMID: 34469738 Free PMC article.
-
Exploiting the Fanconi Anemia Pathway for Targeted Anti-Cancer Therapy.Mol Cells. 2015 Aug;38(8):669-76. doi: 10.14348/molcells.2015.0175. Epub 2015 Jul 21. Mol Cells. 2015. PMID: 26194820 Free PMC article. Review.
-
The multi-functionality of UHRF1: epigenome maintenance and preservation of genome integrity.Nucleic Acids Res. 2021 Jun 21;49(11):6053-6068. doi: 10.1093/nar/gkab293. Nucleic Acids Res. 2021. PMID: 33939809 Free PMC article. Review.
-
SLX4 dampens MutSα-dependent mismatch repair.Nucleic Acids Res. 2022 Mar 21;50(5):2667-2680. doi: 10.1093/nar/gkac075. Nucleic Acids Res. 2022. PMID: 35166826 Free PMC article.
References
-
- Kennedy R.D., D'Andrea A.D. The Fanconi anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi:10.1101/gad.1370505. - DOI - PubMed
-
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007;8:735–748. doi:10.1038/nrg2159. - DOI - PubMed
-
- Collins N., Kupfer G.M. Molecular pathogenesis of Fanconi anemia. Int. J. Hematol. 2005;82:176–183. doi:10.1532/IJH97.05108. - DOI - PubMed
-
- McCabe K.M., Olson S.B., Moses R.E. DNA interstrand crosslink repair in mammalian cells. J. Cell. Physiol. 2009;220:569–573. doi:10.1002/jcp.21811. - DOI - PMC - PubMed
-
- Williams S.A., Kupfer G.M. Ubiquitin and FANC stress responses. In: Bradshaw R.A., Dennis E.A., editors. Handbook of Cell Signaling. 2nd edn. Oxford: Oxford University Press; 2009. pp. 2265–2272.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
