

Understanding the role of TDP-43 and FUS/TLS in ALS and beyond
- PMID: 21813273
- PMCID: PMC3228892
- DOI: 10.1016/j.conb.2011.05.029
Understanding the role of TDP-43 and FUS/TLS in ALS and beyond
Abstract
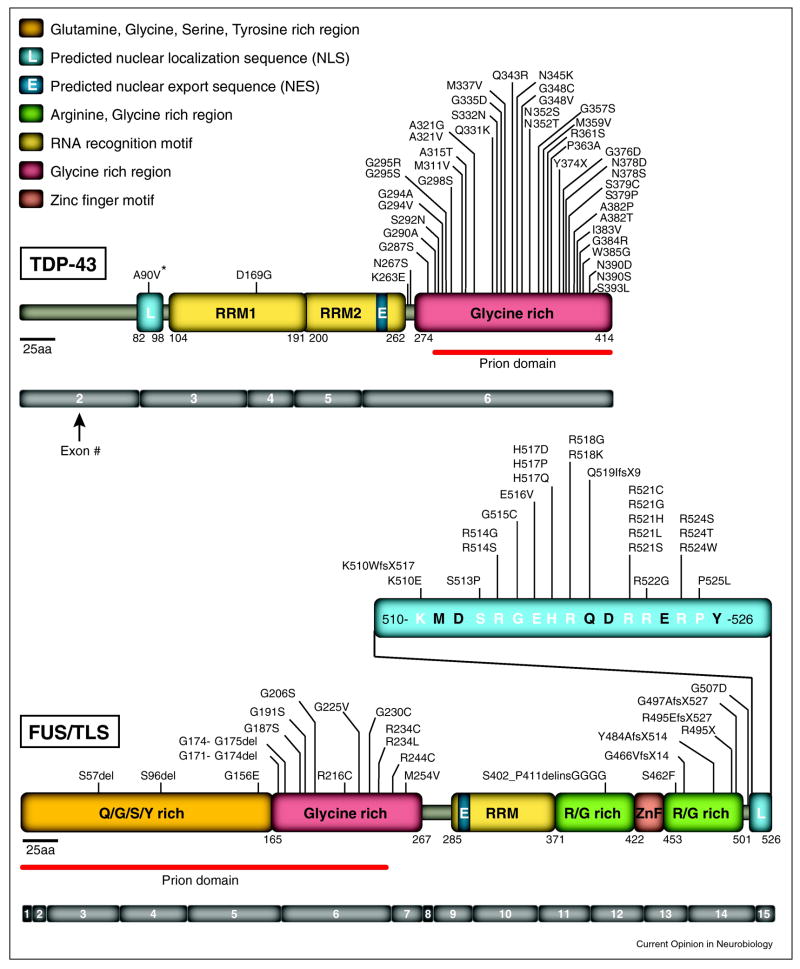
Dominant mutations in two DNA/RNA binding proteins, TDP-43 and FUS/TLS, are causes of inherited Amyotrophic Lateral Sclerosis (ALS). TDP-43 and FUS/TLS have striking structural and functional similarities, implicating alterations in RNA processing as central in ALS. TDP-43 has binding sites within a third of all mouse and human mRNAs in brain and this binding influences the levels and splicing patterns of at least 20% of those mRNAs. Disease modeling in rodents of the first known cause of inherited ALS-mutation in the ubiquitously expressed superoxide dismutase (SOD1)-has yielded non-cell autonomous fatal motor neuron disease caused by one or more toxic properties acquired by the mutant proteins. In contrast, initial disease modeling for TDP-43 and FUS/TLS has produced highly varied phenotypes. It remains unsettled whether TDP-43 and FUS/TLS mutants provoke disease from a loss of function or gain of toxicity or both. TDP-43 or FUS/TLS misaccumulation seems central not just to ALS (where it is found in almost all instances of disease), but more broadly in neurodegenerative disease, including frontal temporal lobular dementia (FTLD-U) and many examples of Alzheimer's or Huntington's disease.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs.Nat Neurosci. 2012 Nov;15(11):1488-97. doi: 10.1038/nn.3230. Epub 2012 Sep 30. Nat Neurosci. 2012. PMID: 23023293 Free PMC article.
-
Rethinking ALS: the FUS about TDP-43.Cell. 2009 Mar 20;136(6):1001-4. doi: 10.1016/j.cell.2009.03.006. Cell. 2009. PMID: 19303844 Free PMC article. Review.
-
ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS.Proc Natl Acad Sci U S A. 2010 Jul 27;107(30):13318-23. doi: 10.1073/pnas.1008227107. Epub 2010 Jul 12. Proc Natl Acad Sci U S A. 2010. PMID: 20624952 Free PMC article.
-
Molecular basis of amyotrophic lateral sclerosis.Prog Neuropsychopharmacol Biol Psychiatry. 2011 Mar 30;35(2):370-2. doi: 10.1016/j.pnpbp.2010.07.017. Epub 2010 Jul 23. Prog Neuropsychopharmacol Biol Psychiatry. 2011. PMID: 20655970 Review.
-
TDP-43/FUS in motor neuron disease: Complexity and challenges.Prog Neurobiol. 2016 Oct-Nov;145-146:78-97. doi: 10.1016/j.pneurobio.2016.09.004. Epub 2016 Sep 28. Prog Neurobiol. 2016. PMID: 27693252 Free PMC article. Review.
Cited by
-
Similar dose-dependence of motor neuron cell death caused by wild type human TDP-43 and mutants with ALS-associated amino acid substitutions.J Biomed Sci. 2013 May 30;20(1):33. doi: 10.1186/1423-0127-20-33. J Biomed Sci. 2013. PMID: 23721326 Free PMC article.
-
The FUS about arginine methylation in ALS and FTLD.EMBO J. 2012 Nov 14;31(22):4249-51. doi: 10.1038/emboj.2012.291. Epub 2012 Oct 19. EMBO J. 2012. PMID: 23085990 Free PMC article.
-
DDX17 is involved in DNA damage repair and modifies FUS toxicity in an RGG-domain dependent manner.Acta Neuropathol. 2021 Sep;142(3):515-536. doi: 10.1007/s00401-021-02333-z. Epub 2021 Jun 1. Acta Neuropathol. 2021. PMID: 34061233 Free PMC article.
-
iPSCs: A Preclinical Drug Research Tool for Neurological Disorders.Int J Mol Sci. 2021 Apr 27;22(9):4596. doi: 10.3390/ijms22094596. Int J Mol Sci. 2021. PMID: 33925625 Free PMC article. Review.
-
Nucleic acid-binding specificity of human FUS protein.Nucleic Acids Res. 2015 Sep 3;43(15):7535-43. doi: 10.1093/nar/gkv679. Epub 2015 Jul 6. Nucleic Acids Res. 2015. PMID: 26150427 Free PMC article.
References
-
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. - PubMed
-
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. - PubMed
-
- Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet. 2010;20:286–293. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
