

Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a 'mitohormesis' mechanism involving reactive oxygen species and PGC-1
- PMID: 21775390
- PMCID: PMC3365271
- DOI: 10.1093/eurheartj/ehr224
Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a 'mitohormesis' mechanism involving reactive oxygen species and PGC-1
Abstract
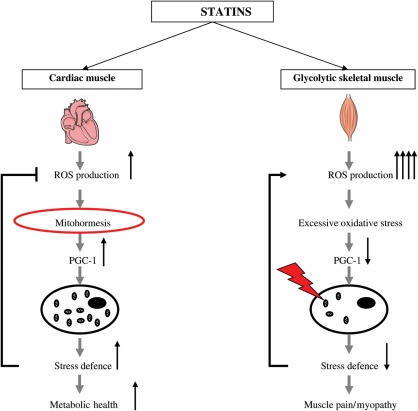
Aims: Statins protect against cardiovascular-related mortality but induce skeletal muscle toxicity. To investigate mechanisms of statins, we tested the hypothesis that statins optimized cardiac mitochondrial function but impaired vulnerable skeletal muscle by inducing different level of reactive oxygen species (ROS).
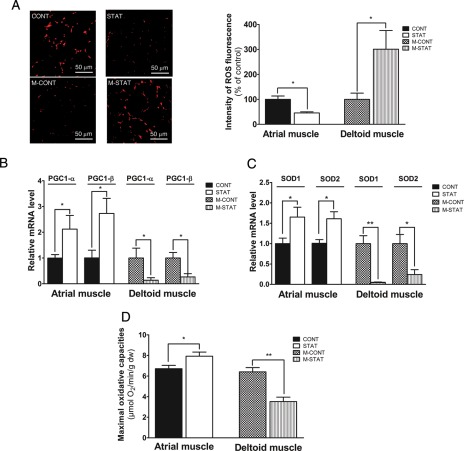
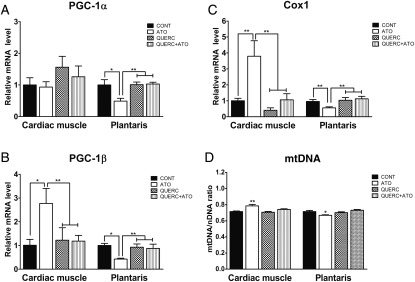
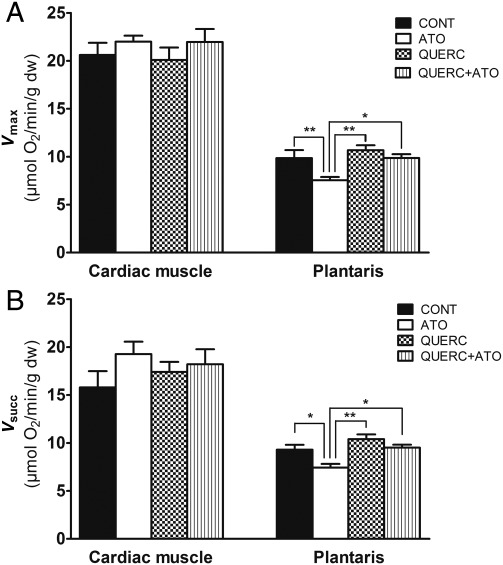
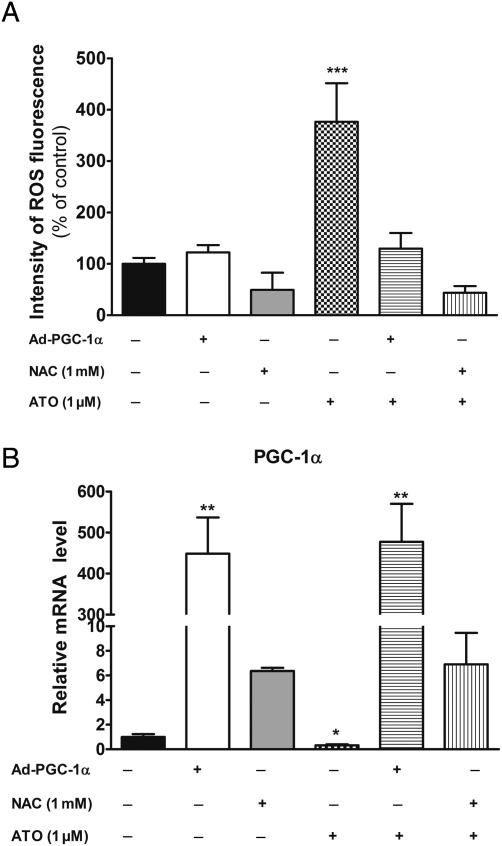
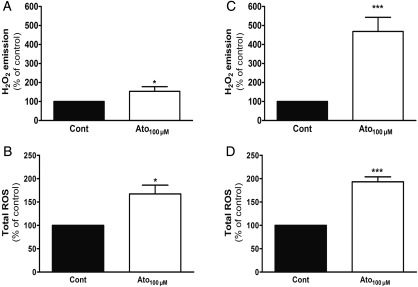
Methods and results: In atrium of patients treated with statins, ROS production was decreased and oxidative capacities were enhanced together with an extensive augmentation of mRNAs expression of peroxisome proliferator-activated receptor gamma co-activator (PGC-1) family. However, in deltoid biopsies from patients with statin-induced muscular myopathy, oxidative capacities were decreased together with ROS increase and a collapse of PGC-1 mRNA expression. Several animal and cell culture experiments were conducted and showed by using ROS scavengers that ROS production was the triggering factor responsible of atorvastatin-induced activation of mitochondrial biogenesis pathway and improvement of antioxidant capacities in heart. Conversely, in skeletal muscle, the large augmentation of ROS production following treatment induced mitochondrial impairments, and reduced mitochondrial biogenesis mechanisms. Quercetin, an antioxidant molecule, was able to counteract skeletal muscle deleterious effects of atorvastatin in rat.
Conclusion: Our findings identify statins as a new activating factor of cardiac mitochondrial biogenesis and antioxidant capacities, and suggest the importance of ROS/PGC-1 signalling pathway as a key element in regulation of mitochondrial function in cardiac as well as skeletal muscles.
Figures


Comment in
-
Mitohormesis: another pleiotropic effect of statins?Eur Heart J. 2012 Jun;33(11):1299-301. doi: 10.1093/eurheartj/ehr287. Epub 2011 Oct 6. Eur Heart J. 2012. PMID: 21979988 Free PMC article. No abstract available.
Similar articles
-
Mitohormesis: another pleiotropic effect of statins?Eur Heart J. 2012 Jun;33(11):1299-301. doi: 10.1093/eurheartj/ehr287. Epub 2011 Oct 6. Eur Heart J. 2012. PMID: 21979988 Free PMC article. No abstract available.
-
PGC-1β modulates statin-associated myotoxicity in mice.Arch Toxicol. 2019 Feb;93(2):487-504. doi: 10.1007/s00204-018-2369-7. Epub 2018 Dec 3. Arch Toxicol. 2019. PMID: 30511338
-
Mitochondria of trained skeletal muscle are protected from deleterious effects of statins.Muscle Nerve. 2012 Sep;46(3):367-73. doi: 10.1002/mus.23309. Muscle Nerve. 2012. PMID: 22907227
-
The role of mitochondria in statin-induced myopathy.Eur J Clin Invest. 2015 Jul;45(7):745-54. doi: 10.1111/eci.12461. Epub 2015 Jun 15. Eur J Clin Invest. 2015. PMID: 25991405 Review.
-
Do antioxidant supplements interfere with skeletal muscle adaptation to exercise training?J Physiol. 2016 Sep 15;594(18):5135-47. doi: 10.1113/JP270654. Epub 2016 Jan 18. J Physiol. 2016. PMID: 26638792 Free PMC article. Review.
Cited by
-
Skeletal muscle mitochondrial dysfunction precedes right ventricular impairment in experimental pulmonary hypertension.Mol Cell Biochem. 2013 Jan;373(1-2):161-70. doi: 10.1007/s11010-012-1485-6. Epub 2012 Oct 26. Mol Cell Biochem. 2013. PMID: 23099843
-
Effects of Simvastatin on Lipid Metabolism in Wild-Type Mice and Mice with Muscle PGC-1α Overexpression.Int J Mol Sci. 2021 May 7;22(9):4950. doi: 10.3390/ijms22094950. Int J Mol Sci. 2021. PMID: 34066911 Free PMC article.
-
Statin-related Muscle Toxicity: An Evidence-based Review.touchREV Endocrinol. 2022 Nov;18(2):89-95. doi: 10.17925/EE.2022.18.2.89. Epub 2022 Nov 21. touchREV Endocrinol. 2022. PMID: 36694885 Free PMC article. Review.
-
Statins in High Cardiovascular Risk Patients: Do Comorbidities and Characteristics Matter?Int J Mol Sci. 2022 Aug 18;23(16):9326. doi: 10.3390/ijms23169326. Int J Mol Sci. 2022. PMID: 36012589 Free PMC article. Review.
-
Effects of exercise training on mitochondrial function in patients with type 2 diabetes.World J Diabetes. 2014 Aug 15;5(4):482-92. doi: 10.4239/wjd.v5.i4.482. World J Diabetes. 2014. PMID: 25126394 Free PMC article. Review.
References
-
- Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346:539–540. doi:10.1056/NEJM200202143460721. - DOI - PubMed
-
- Jasinska M, Owczarek J, Orszulak-Michalak D. Statins: a new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol Rep. 2007;59:483–499. - PubMed
-
- Giordano N, Senesi M, Mattii G, Battisti E, Villanova M, Gennari C. Polymyositis associated with simvastatin. Lancet. 1997;349:1600–1601. doi:10.1016/S0140-6736(05)61628-5. - DOI - PubMed
-
- Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering—are they clinically relevant? Eur Heart J. 2003;24:225–248. doi:10.1016/S0195-668X(02)00419-0. - DOI - PubMed
-
- Echaniz-Laguna A, Mohr M, Tranchant C. Neuromuscular symptoms and elevated creatine kinase after statin withdrawal. N Engl J Med. 2010;362:564–565. doi:10.1056/NEJMc0908215. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
