

Faster SEQUEST searching for peptide identification from tandem mass spectra
- PMID: 21761931
- PMCID: PMC3166376
- DOI: 10.1021/pr101196n
Faster SEQUEST searching for peptide identification from tandem mass spectra
Abstract
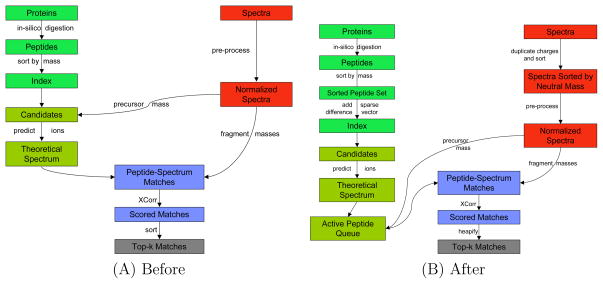
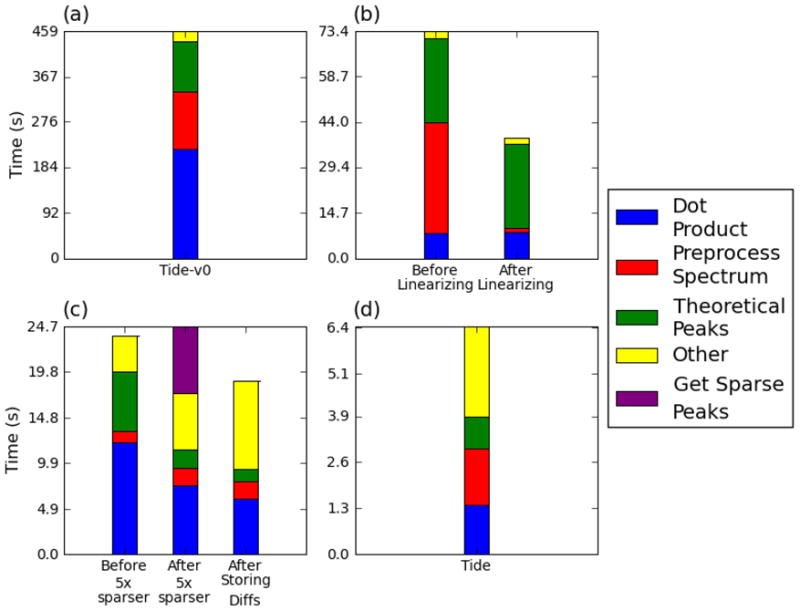
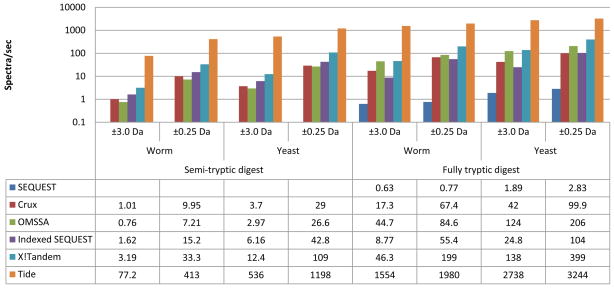
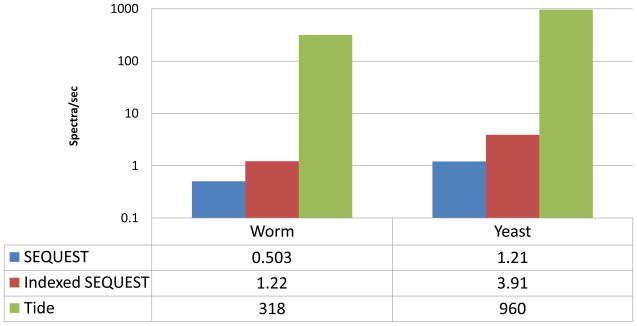
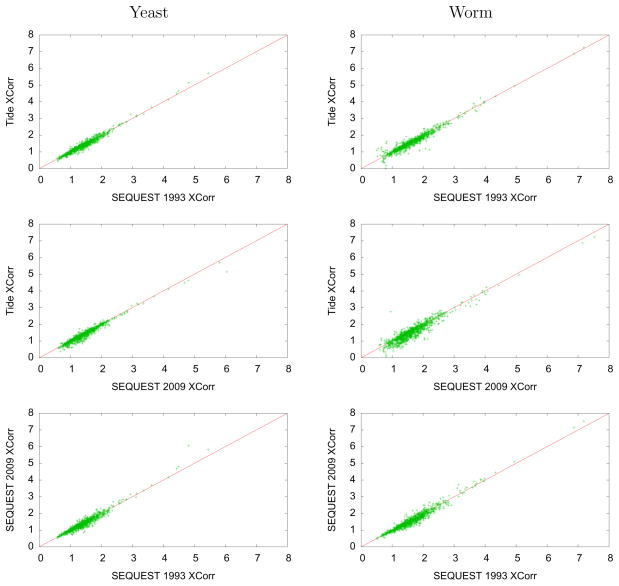
Computational analysis of mass spectra remains the bottleneck in many proteomics experiments. SEQUEST was one of the earliest software packages to identify peptides from mass spectra by searching a database of known peptides. Though still popular, SEQUEST performs slowly. Crux and TurboSEQUEST have successfully sped up SEQUEST by adding a precomputed index to the search, but the demand for ever-faster peptide identification software continues to grow. Tide, introduced here, is a software program that implements the SEQUEST algorithm for peptide identification and that achieves a dramatic speedup over Crux and SEQUEST. The optimization strategies detailed here employ a combination of algorithmic and software engineering techniques to achieve speeds up to 170 times faster than a recent version of SEQUEST that uses indexing. For example, on a single Xeon CPU, Tide searches 10,000 spectra against a tryptic database of 27,499 Caenorhabditis elegans proteins at a rate of 1550 spectra per second, which compares favorably with a rate of 8.8 spectra per second for a recent version of SEQUEST with index running on the same hardware.
Figures
Similar articles
-
ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity.J Proteomics. 2015 Nov 3;129:16-24. doi: 10.1016/j.jprot.2015.07.001. Epub 2015 Jul 11. J Proteomics. 2015. PMID: 26171723 Free PMC article.
-
Speeding up tandem mass spectrometry based database searching by peptide and spectrum indexing.Rapid Commun Mass Spectrom. 2010 Mar;24(6):807-14. doi: 10.1002/rcm.4448. Rapid Commun Mass Spectrom. 2010. PMID: 20187083
-
Optimization of filtering criterion for SEQUEST database searching to improve proteome coverage in shotgun proteomics.BMC Bioinformatics. 2007 Aug 31;8:323. doi: 10.1186/1471-2105-8-323. BMC Bioinformatics. 2007. PMID: 17761002 Free PMC article.
-
Protein identification using Sorcerer 2 and SEQUEST.Curr Protoc Bioinformatics. 2009 Dec;Chapter 13:Unit 13.3. doi: 10.1002/0471250953.bi1303s28. Curr Protoc Bioinformatics. 2009. PMID: 19957274 Review.
-
The spectral networks paradigm in high throughput mass spectrometry.Mol Biosyst. 2012 Oct;8(10):2535-44. doi: 10.1039/c2mb25085c. Mol Biosyst. 2012. PMID: 22610447 Free PMC article. Review.
Cited by
-
The SEQUEST family tree.J Am Soc Mass Spectrom. 2015 Nov;26(11):1814-9. doi: 10.1007/s13361-015-1201-3. Epub 2015 Jun 30. J Am Soc Mass Spectrom. 2015. PMID: 26122518 Free PMC article.
-
Bioinformatic Workflows for Metaproteomics.Methods Mol Biol. 2024;2820:187-213. doi: 10.1007/978-1-0716-3910-8_16. Methods Mol Biol. 2024. PMID: 38941024
-
Schistosoma haematobium Extracellular Vesicle Proteins Confer Protection in a Heterologous Model of Schistosomiasis.Vaccines (Basel). 2020 Jul 24;8(3):416. doi: 10.3390/vaccines8030416. Vaccines (Basel). 2020. PMID: 32722279 Free PMC article.
-
ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity.J Proteomics. 2015 Nov 3;129:16-24. doi: 10.1016/j.jprot.2015.07.001. Epub 2015 Jul 11. J Proteomics. 2015. PMID: 26171723 Free PMC article.
-
PeptideShaker Online: A User-Friendly Web-Based Framework for the Identification of Mass Spectrometry-Based Proteomics Data.J Proteome Res. 2021 Dec 3;20(12):5419-5423. doi: 10.1021/acs.jproteome.1c00678. Epub 2021 Oct 28. J Proteome Res. 2021. PMID: 34709836 Free PMC article.
References
-
- Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. - PubMed
-
- Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nature Methods. 2007;4(10):787–797. - PubMed
-
- Eng JK, Fischer B, Grossman J, MacCoss MJ. A fast SEQUEST cross correlation algorithm. Journal of Proteome Research. 2008;7(10):4598–4602. - PubMed
-
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
