

Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia
- PMID: 21752874
- PMCID: PMC3141878
- DOI: 10.1124/pr.110.004135
Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia
Abstract
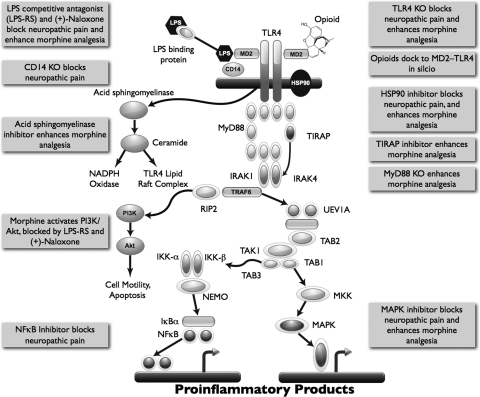
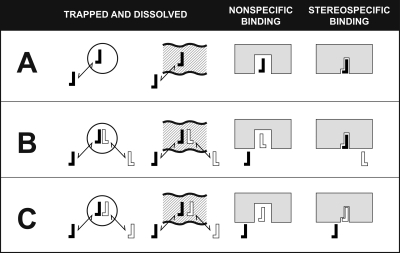
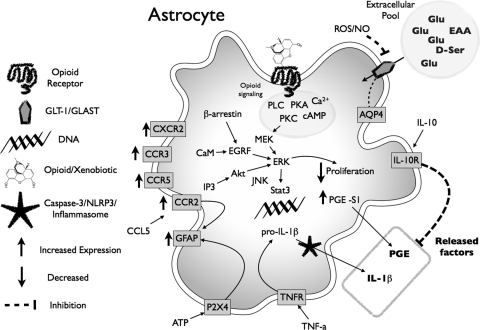
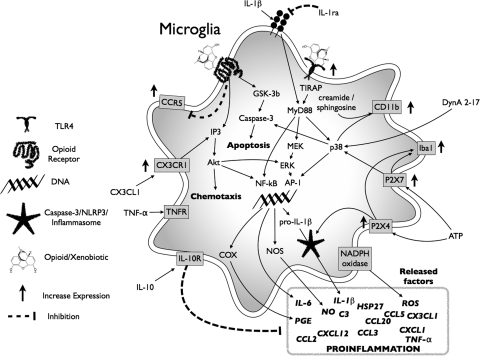
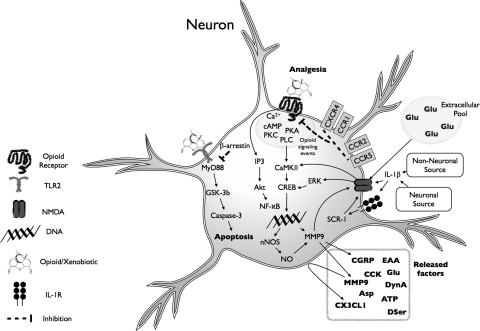
Vastly stimulated by the discovery of opioid receptors in the early 1970s, preclinical and clinical research was directed at the study of stereoselective neuronal actions of opioids, especially those played in their crucial analgesic role. However, during the past decade, a new appreciation of the non-neuronal actions of opioids has emerged from preclinical research, with specific appreciation for the nonclassic and nonstereoselective sites of action. Opioid activity at Toll-like receptors, newly recognized innate immune pattern recognition receptors, adds substantially to this unfolding story. It is now apparent from molecular and rodent data that these newly identified signaling events significantly modify the pharmacodynamics of opioids by eliciting proinflammatory reactivity from glia, the immunocompetent cells of the central nervous system. These central immune signaling events, including the release of cytokines and chemokines and the associated disruption of glutamate homeostasis, cause elevated neuronal excitability, which subsequently decreases opioid analgesic efficacy and leads to heightened pain states. This review will examine the current preclinical literature of opioid-induced central immune signaling mediated by classic and nonclassic opioid receptors. A unification of the preclinical pharmacology, neuroscience, and immunology of opioids now provides new insights into common mechanisms of chronic pain, naive tolerance, analgesic tolerance, opioid-induced hyperalgesia, and allodynia. Novel pharmacological targets for future drug development are discussed in the hope that disease-modifying chronic pain treatments arising from the appreciation of opioid-induced central immune signaling may become practical.
Figures
Similar articles
-
Opioid-induced central immune signaling: implications for opioid analgesia.Headache. 2015 Apr;55(4):475-89. doi: 10.1111/head.12552. Epub 2015 Mar 31. Headache. 2015. PMID: 25833219 Free PMC article. Review.
-
Exploring neuroinflammation as a potential avenue to improve the clinical efficacy of opioids.Expert Rev Neurother. 2012 Nov;12(11):1311-24. doi: 10.1586/ern.12.125. Expert Rev Neurother. 2012. PMID: 23234393 Review.
-
Targeting metabotropic glutamate receptors in neuroimmune communication.Neuropharmacology. 2012 Sep;63(4):501-6. doi: 10.1016/j.neuropharm.2012.05.024. Epub 2012 May 26. Neuropharmacology. 2012. PMID: 22640632 Review.
-
Toll-Like Receptor 4 (TLR4)/Opioid Receptor Pathway Crosstalk and Impact on Opioid Analgesia, Immune Function, and Gastrointestinal Motility.Front Immunol. 2020 Jul 8;11:1455. doi: 10.3389/fimmu.2020.01455. eCollection 2020. Front Immunol. 2020. PMID: 32733481 Free PMC article. Review.
-
Opioid Receptors.Annu Rev Med. 2016;67:433-51. doi: 10.1146/annurev-med-062613-093100. Epub 2015 Aug 26. Annu Rev Med. 2016. PMID: 26332001 Review.
Cited by
-
Immune response and cytokine profiles in post-laminectomy pain syndrome: comparative analysis after treatment with intrathecal opioids, oral opioids, and non-opioid therapies.Inflammopharmacology. 2024 Oct;32(5):3295-3309. doi: 10.1007/s10787-024-01521-z. Epub 2024 Jul 22. Inflammopharmacology. 2024. PMID: 39039349
-
Morphine amplifies mechanical allodynia via TLR4 in a rat model of spinal cord injury.Brain Behav Immun. 2016 Nov;58:348-356. doi: 10.1016/j.bbi.2016.08.004. Epub 2016 Aug 9. Brain Behav Immun. 2016. PMID: 27519154 Free PMC article.
-
Challenges for opioid receptor nomenclature: IUPHAR Review 9.Br J Pharmacol. 2015 Jan;172(2):317-23. doi: 10.1111/bph.12612. Epub 2014 Jul 1. Br J Pharmacol. 2015. PMID: 24528283 Free PMC article. Review.
-
Oxycodone, fentanyl, and morphine amplify established neuropathic pain in male rats.Pain. 2019 Nov;160(11):2634-2640. doi: 10.1097/j.pain.0000000000001652. Pain. 2019. PMID: 31299018 Free PMC article.
-
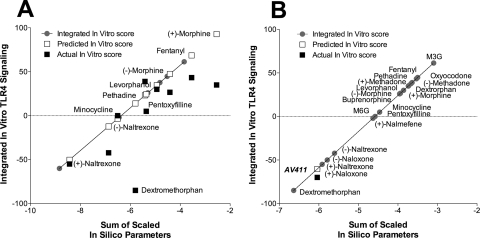
Systemic administration of propentofylline, ibudilast, and (+)-naltrexone each reverses mechanical allodynia in a novel rat model of central neuropathic pain.J Pain. 2014 Apr;15(4):407-21. doi: 10.1016/j.jpain.2013.12.007. Epub 2014 Jan 9. J Pain. 2014. PMID: 24412802 Free PMC article.
References
-
- Adler MW, Rogers TJ. (2005) Are chemokines the third major system in the brain? J Leukoc Biol 78:1204–1209 - PubMed
-
- Agostini S, Eutamene H, Cartier C, Broccardo M, Improta G, Houdeau E, Petrella C, Ferrier L, Theodorou V, Bueno L. (2010) Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 139:553–563, 563.e551–555 - PubMed
-
- Alonso E, Garrido E, Díez-Fernández C, Pérez-García C, Herradón G, Ezquerra L, Deuel TF, Alguacil LF. (2007) Yohimbine prevents morphine-induced changes of glial fibrillary acidic protein in brainstem and alpha2-adrenoceptor gene expression in hippocampus. Neurosci Lett 412:163–167 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous