

AlignHUSH: alignment of HMMs using structure and hydrophobicity information
- PMID: 21729312
- PMCID: PMC3228556
- DOI: 10.1186/1471-2105-12-275
AlignHUSH: alignment of HMMs using structure and hydrophobicity information
Abstract
Background: Sensitive remote homology detection and accurate alignments especially in the midnight zone of sequence similarity are needed for better function annotation and structural modeling of proteins. An algorithm, AlignHUSH for HMM-HMM alignment has been developed which is capable of recognizing distantly related domain families The method uses structural information, in the form of predicted secondary structure probabilities, and hydrophobicity of amino acids to align HMMs of two sets of aligned sequences. The effect of using adjoining column(s) information has also been investigated and is found to increase the sensitivity of HMM-HMM alignments and remote homology detection.
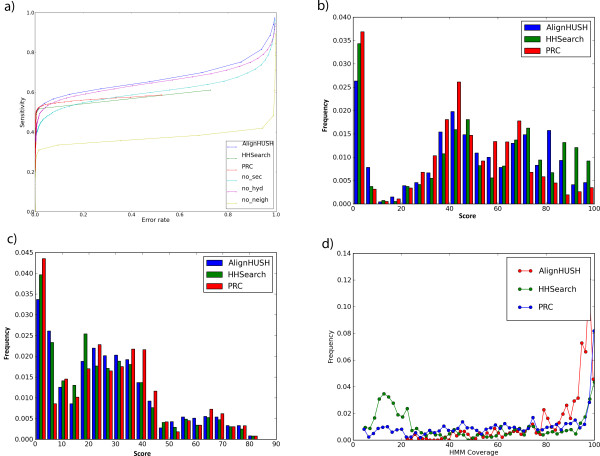
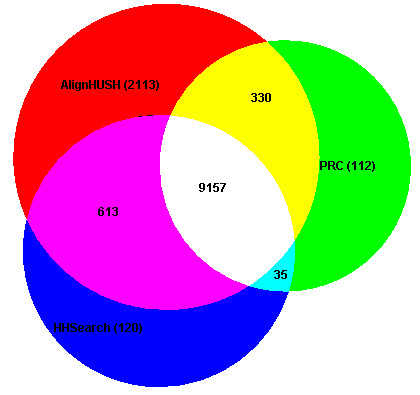
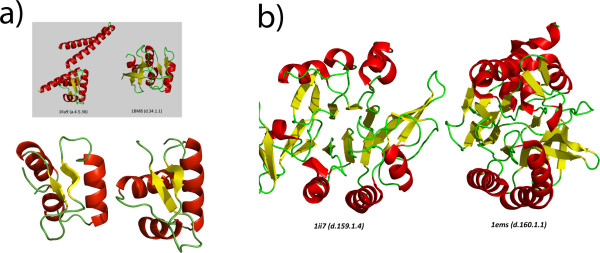
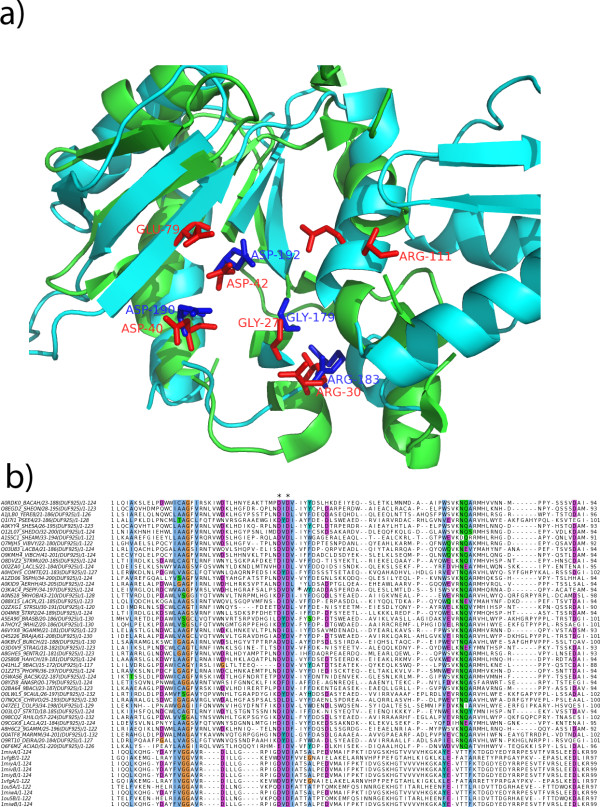
Results: We have assessed the performance of AlignHUSH using known evolutionary relationships available in SCOP. AlignHUSH performs better than the best HMM-HMM alignment methods and is observed to be even more sensitive at higher error rates. Accuracy of the alignments obtained using AlignHUSH has been assessed using the structure-based alignments available in BaliBASE. The alignment length and the alignment quality are found to be appropriate for homology modeling and function annotation. The alignment accuracy is found to be comparable to existing methods for profile-profile alignments.
Conclusions: A new method to align HMMs has been developed and is shown to have better sensitivity at error rates of 10% and above when compared to other available programs. The proposed method could effectively aid obtaining clues to functions of proteins of yet unknown function. A web-server incorporating the AlignHUSH method is available at http://crick.mbu.iisc.ernet.in/~alignhush/
Figures
Similar articles
-
Alignment of multiple proteins with an ensemble of hidden Markov models.Int J Data Min Bioinform. 2010;4(1):60-71. doi: 10.1504/ijdmb.2010.030967. Int J Data Min Bioinform. 2010. PMID: 20376922 Free PMC article.
-
Protein homology detection by HMM-HMM comparison.Bioinformatics. 2005 Apr 1;21(7):951-60. doi: 10.1093/bioinformatics/bti125. Epub 2004 Nov 5. Bioinformatics. 2005. PMID: 15531603
-
HMM-ModE--improved classification using profile hidden Markov models by optimising the discrimination threshold and modifying emission probabilities with negative training sequences.BMC Bioinformatics. 2007 Mar 27;8:104. doi: 10.1186/1471-2105-8-104. BMC Bioinformatics. 2007. PMID: 17389042 Free PMC article.
-
Five hierarchical levels of sequence-structure correlation in proteins.Appl Bioinformatics. 2004;3(2-3):97-104. doi: 10.2165/00822942-200403020-00004. Appl Bioinformatics. 2004. PMID: 15693735 Review.
-
Sequence and structure alignments in post-AlphaFold era.Curr Opin Struct Biol. 2023 Apr;79:102539. doi: 10.1016/j.sbi.2023.102539. Epub 2023 Feb 6. Curr Opin Struct Biol. 2023. PMID: 36753924 Review.
Cited by
-
Accelerating Information Retrieval from Profile Hidden Markov Model Databases.PLoS One. 2016 Nov 22;11(11):e0166358. doi: 10.1371/journal.pone.0166358. eCollection 2016. PLoS One. 2016. PMID: 27875548 Free PMC article.
-
De-DUFing the DUFs: Deciphering distant evolutionary relationships of Domains of Unknown Function using sensitive homology detection methods.Biol Direct. 2015 Jul 31;10:38. doi: 10.1186/s13062-015-0069-2. Biol Direct. 2015. PMID: 26228684 Free PMC article.
-
Computational recognition and analysis of hitherto uncharacterized nucleotide cyclase-like proteins in bacteria.Biol Direct. 2016 May 31;11:27. doi: 10.1186/s13062-016-0130-9. Biol Direct. 2016. PMID: 27246835 Free PMC article.
-
Using HHsearch to tackle proteins of unknown function: A pilot study with PH domains.Traffic. 2016 Nov;17(11):1214-1226. doi: 10.1111/tra.12432. Epub 2016 Oct 9. Traffic. 2016. PMID: 27601190 Free PMC article.
-
Genome-wide search for eliminylating domains reveals novel function for BLES03-like proteins.Genome Biol Evol. 2014 Jul 24;6(8):2017-33. doi: 10.1093/gbe/evu161. Genome Biol Evol. 2014. PMID: 25062915 Free PMC article.
References
-
- Bhadra R, Srinivasan N, Pandit SB. A new domain family in the superfamily of alkaline phosphatases. In Silico Biol. 2005;5:379–387. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
