

Review
doi: 10.1084/jem.20110551.
Integrating mechanisms of pulmonary fibrosis
Affiliations
- PMID: 21727191
- PMCID: PMC3136685
- DOI: 10.1084/jem.20110551
Item in Clipboard
Review
Integrating mechanisms of pulmonary fibrosis
J Exp Med.
.
Abstract
Pulmonary fibrosis is a highly heterogeneous and lethal pathological process with limited therapeutic options. Although research on the pathogenesis of pulmonary fibrosis has frequently focused on the mechanisms that regulate the proliferation, activation, and differentiation of collagen-secreting myofibroblasts, recent studies have identified new pathogenic mechanisms that are critically involved in the initiation and progression of fibrosis in a variety of settings. A more detailed and integrated understanding of the cellular and molecular mechanisms of pulmonary fibrosis could help pave the way for effective therapeutics for this devastating and complex disease.
Figures
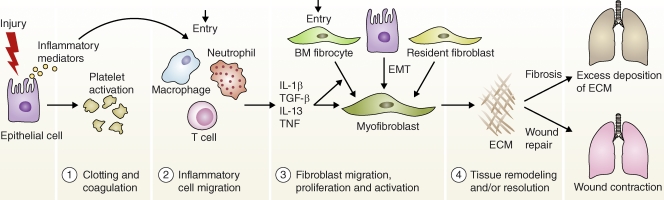
Disruptions in normal wound healing contribute to the development of pulmonary fibrosis. Wound healing has four distinct stages: a clotting/coagulation phase (1), an inflammatory cell migration phase (2), a fibroblast migration/proliferation/activation phase (3), and a tissue remodeling and resolution phase (4). After lung injury, epithelial cells release inflammatory mediators that initiate an antifibrinolytic coagulation cascade, which triggers platelet activation and blood clot formation. This is followed by entry of leukocytes (e.g., neutrophils, macrophages, and T cells). The recruited leukocytes secrete profibrotic cytokines such as IL-1β, TNF, IL-13, and TGF-β. The activated macrophages and neutrophils also remove dead cells and eliminate any invading organisms. In the subsequent phase, fibrocytes from the bone marrow and resident fibroblasts proliferate and differentiate into myofibroblasts, which release ECM components. Fibroblasts and myofibroblasts may also be derived from epithelial cells undergoing EMT. In the final remodeling and resolution phase, activated myofibroblasts can promote wound repair, leading to wound contraction and restoration of blood vessels. However, fibrosis often develops if any stage in the tissue repair program is dysregulated or when the lung-damaging stimulus persists.
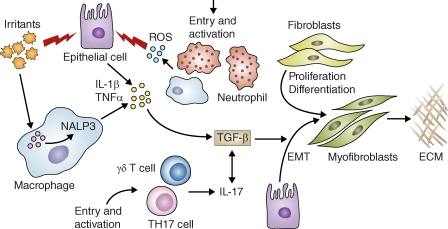
Proinflammatory and profibrotic mediators in the initiation and maintenance of fibrosis. Irritants like silica, asbestos, and bleomycin (uric acid) can injure lung epithelial cells and can be detected by the Nalp3 inflammasome in macrophages. These irritants stimulate the production of ROS, chemokines, and cytokines. These inflammatory mediators enhance the recruitment and activation of leukocytes at the site of tissue injury. For example, IL-1β induces the activation of ROS-expressing neutrophils, which can further damage epithelial cells. IL-1β also promotes production of TGF-β1, an important profibrotic cytokine that triggers fibroblast proliferation and activation. TGF-β also targets epithelial cells, inducing EMT and the formation of ECM-producing myofibroblasts. TGF-β1 further exacerbates the inflammatory response by stimulating the differentiation of Th17 cells. Interactive PPT slides for this figure are available online.
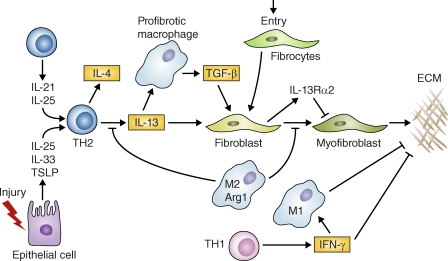
Specialized subsets of T helper cells and macrophages play distinct roles in pulmonary fibrosis. After injury, epithelial cells release IL-25, IL-33, and TSLP, which can facilitate the development of profibrotic Th2 responses. T cells also release IL-21 and IL-25, which promote Th2 differentiation. Th2 cells release IL-4 and IL-13, which promote the development of a profibrotic macrophage subpopulation that secretes TGF-β1 among other mediators. IL-13 can also directly activate fibroblasts independently of TGF-β1. Th2 cytokines also trigger specific chemokines that promote the recruitment of collagen-secreting fibrocytes from the bone marrow, which amplify fibrotic responses. The resulting myofibroblasts release ECM components. However, Th2 cytokines can also trigger antifibrotic feedback mechanisms. For example, Th2 cytokines activate arginase-1 activity in M2 macrophages, which inhibit further IL-13 production and myofibroblast differentiation. IL-13 can also up-regulate the IL-13 decoy receptor in fibroblasts, which antagonizes ECM production via a negative feedback loop. In addition, IFN-γ, produced by Th1 cells, exhibits potent antifibrotic activity by suppressing collagen synthesis in fibroblasts and by promoting the activation of inflammatory M1 macrophages that favor ECM degradation. Interactive PPT slides for this figure are available online.
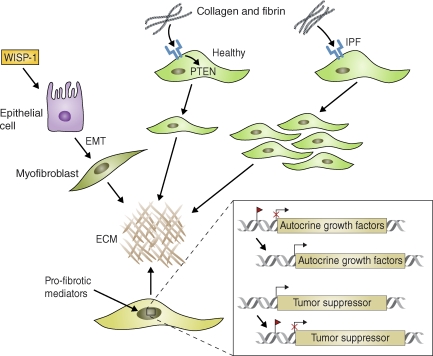
Intrinsic changes in the activation status of epithelial cells and fibroblasts can promote growth factor–independent pulmonary fibrosis. Wnt–β-catenin signaling activated (for example) by WISP-1, is constitutively active in some ATII epithelial cells in IPF patients and in mice with bleomycin-induced pulmonary fibrosis. This signaling triggers EMT and synthesis of ECM components by fibroblasts. In healthy fibroblasts, collagen-mediated stimulation of β1 integrin (blue) up-regulates PTEN activity and inhibits proliferation. IPF fibroblasts, however, display a pathological pattern of β1 integrin expression and signaling that can lead to decreased PTEN expression, aberrant activation of PI3 kinase, and excessive proliferation. Profibrotic mediators also promote epigenetic changes in fibroblasts that contribute to the pathogenesis of fibrosis. For example, the promoter regions of various genes encoding autocrine growth and/or differentiation factors can be demethylated, leading to their sustained and heritable activation. In addition, tumor suppressor genes can become methylated (red flag) and therefore inactivated, leading to the sustained activation of oncogenes that promote growth factor–independent proliferation of fibroblasts. miRNAs (e.g., miR-21) may operate in a similar fashion by blocking the translation or promoting the degradation of tumor suppressor genes in fibroblasts.
Similar articles
-
Cellular players in lung fibrosis.Curr Pharm Des. 2012;18(27):4093-102. doi: 10.2174/138161212802430396. Curr Pharm Des. 2012. PMID: 22630084 Review.
-
Molecular mechanisms of pulmonary fibrosis and current treatment.Curr Mol Med. 2001 Nov;1(5):551-73. doi: 10.2174/1566524013363401. Curr Mol Med. 2001. PMID: 11899231 Review.
-
The role of TGF-beta in pulmonary fibrosis.Ciba Found Symp. 1991;157:194-207; discussion 207-11. doi: 10.1002/9780470514061.ch13. Ciba Found Symp. 1991. PMID: 1712697 Review.
-
The role of circulating mesenchymal progenitor cells, fibrocytes, in promoting pulmonary fibrosis.Trans Am Clin Climatol Assoc. 2009;120:49-59. Trans Am Clin Climatol Assoc. 2009. PMID: 19768162 Free PMC article. Review.
-
Molecular mechanisms of and possible treatment strategies for idiopathic pulmonary fibrosis.Curr Pharm Des. 2005;11(30):3943-71. doi: 10.2174/138161205774580561. Curr Pharm Des. 2005. PMID: 16305523 Review.
Cited by
-
Safety pharmacology of self-assembled-micelle inhibitory RNA-targeting amphiregulin (SAMiRNA-AREG), a novel siRNA nanoparticle platform.Toxicol Rep. 2021 Mar 31;8:839-845. doi: 10.1016/j.toxrep.2021.03.022. eCollection 2021. Toxicol Rep. 2021. PMID: 33912399 Free PMC article.
-
The Effect of Nintedanib on T-Cell Activation, Subsets and Functions.Drug Des Devel Ther. 2021 Mar 8;15:997-1011. doi: 10.2147/DDDT.S288369. eCollection 2021. Drug Des Devel Ther. 2021. PMID: 33727792 Free PMC article.
-
Targeting the AXL Receptor in Combating Smoking-related Pulmonary Fibrosis.Am J Respir Cell Mol Biol. 2021 Jun;64(6):734-746. doi: 10.1165/rcmb.2020-0303OC. Am J Respir Cell Mol Biol. 2021. PMID: 33730527 Free PMC article.
-
The impact of the myeloid response to radiation therapy.Clin Dev Immunol. 2013;2013:281958. doi: 10.1155/2013/281958. Epub 2013 Apr 7. Clin Dev Immunol. 2013. PMID: 23653658 Free PMC article. Review.
-
Delivery of RNAi Therapeutics to the Airways-From Bench to Bedside.Molecules. 2016 Sep 20;21(9):1249. doi: 10.3390/molecules21091249. Molecules. 2016. PMID: 27657028 Free PMC article. Review.
References
-
- Asangani I.A., Rasheed S.A., Nikolova D.A., Leupold J.H., Colburn N.H., Post S., Allgayer H. 2008. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 27:2128–2136 10.1038/sj.onc.1210856 - DOI - PubMed
-
- Atabai K., Jame S., Azhar N., Kuo A., Lam M., McKleroy W., Dehart G., Rahman S., Xia D.D., Melton A.C., et al. 2009. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Invest. 119:3713–3722 10.1172/JCI40053 - DOI - PMC - PubMed
-
- Bauman K.A., Wettlaufer S.H., Okunishi K., Vannella K.M., Stoolman J.S., Huang S.K., Courey A.J., White E.S., Hogaboam C.M., Simon R.H., et al. 2010. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Invest. 120:1950–1960 10.1172/JCI38369 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
