

Golgi glycosylation and human inherited diseases
- PMID: 21709180
- PMCID: PMC3181031
- DOI: 10.1101/cshperspect.a005371
Golgi glycosylation and human inherited diseases
Abstract
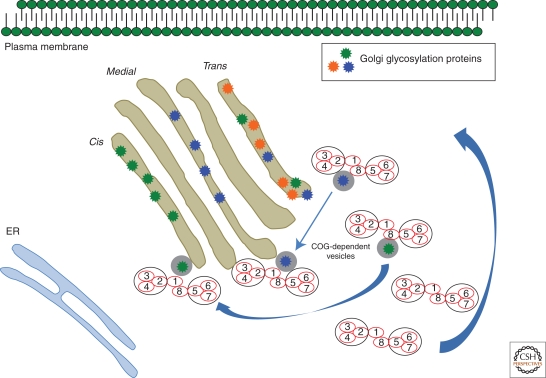
The Golgi factory receives custom glycosylates and dispatches its cargo to the correct cellular locations. The process requires importing donor substrates, moving the cargo, and recycling machinery. Correctly glycosylated cargo reflects the Golgi's quality and efficiency. Genetic disorders in the specific equipment (enzymes), donors (nucleotide sugar transporters), or equipment recycling/reorganization components (COG, SEC, golgins) can all affect glycosylation. Dozens of human glycosylation disorders fit these categories. Many other genes, with or without familiar names, well-annotated pedigrees, or likely homologies will join the ranks of glycosylation disorders. Their broad and unpredictable case-by-case phenotypes cross the traditional medical specialty boundaries. The gene functions in patients may be elusive, but their common feature may include altered glycosylation that provide clues to Golgi function. This article focuses on a group of human disorders that affect protein or lipid glycosylation. Readers may find it useful to generalize some of these patient-based, translational observations to their own research.
Figures
Similar articles
-
Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review.Clin Chem. 2006 Apr;52(4):574-600. doi: 10.1373/clinchem.2005.063040. Epub 2006 Feb 23. Clin Chem. 2006. PMID: 16497938 Review.
-
Genetic defects in the human glycome.Nat Rev Genet. 2006 Jul;7(7):537-51. doi: 10.1038/nrg1894. Epub 2006 Jun 6. Nat Rev Genet. 2006. PMID: 16755287 Review.
-
Glycosylation Quality Control by the Golgi Structure.J Mol Biol. 2016 Aug 14;428(16):3183-3193. doi: 10.1016/j.jmb.2016.02.030. Epub 2016 Mar 5. J Mol Biol. 2016. PMID: 26956395 Free PMC article. Review.
-
Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation.Carbohydr Res. 2008 Aug 11;343(12):2024-31. doi: 10.1016/j.carres.2008.01.034. Epub 2008 Feb 2. Carbohydr Res. 2008. PMID: 18353293 Free PMC article. Review.
-
How Golgi glycosylation meets and needs trafficking: the case of the COG complex.Glycobiology. 2011 Jul;21(7):853-63. doi: 10.1093/glycob/cwq179. Epub 2010 Nov 26. Glycobiology. 2011. PMID: 21112967 Review.
Cited by
-
Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation.Int J Mol Sci. 2020 Jun 30;21(13):4654. doi: 10.3390/ijms21134654. Int J Mol Sci. 2020. PMID: 32629928 Free PMC article. Review.
-
Golgi compartments enable controlled biomolecular assembly using promiscuous enzymes.Elife. 2020 Jun 29;9:e49573. doi: 10.7554/eLife.49573. Elife. 2020. PMID: 32597757 Free PMC article.
-
Fractionated plasma N-glycan profiling of novel cohort of ATP6AP1-CDG subjects identifies phenotypic association.J Inherit Metab Dis. 2023 Mar;46(2):300-312. doi: 10.1002/jimd.12589. Epub 2023 Jan 29. J Inherit Metab Dis. 2023. PMID: 36651831 Free PMC article.
-
Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster.Front Genet. 2018 Oct 2;9:436. doi: 10.3389/fgene.2018.00436. eCollection 2018. Front Genet. 2018. PMID: 30333856 Free PMC article. Review.
-
Dissecting functions of the conserved oligomeric Golgi tethering complex using a cell-free assay.Traffic. 2014 Jan;15(1):12-21. doi: 10.1111/tra.12128. Epub 2013 Oct 31. Traffic. 2014. PMID: 24102787 Free PMC article.
References
-
- Akama TO, Nishida K, Nakayama J, Watanabe H, Ozaki K, Nakamura T, Dota A, Kawasaki S, Inoue Y, Maeda N, et al. 2000. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet 26: 237–241 - PubMed
-
- Al-Dosari M, Alkuraya FS 2009. A novel missense mutation in SCYL1BP1 produces geroderma osteodysplastica phenotype indistinguishable from that caused by nullimorphic mutations. Am J Med Genet A 149A: 2093–2098 - PubMed
-
- Al-Gazali LI, Sztriha L, Skaff F, Haas D 2001. Gerodermia osteodysplastica and wrinkly skin syndrome: Are they the same? Am J Med Genet 101: 213–220 - PubMed
-
- Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, Richards S, Patterson S, Kotsianidis I, Mollica L, et al. 2006. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 12: 846–851 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical