

The reconstruction of condition-specific transcriptional modules provides new insights in the evolution of yeast AP-1 proteins
- PMID: 21695268
- PMCID: PMC3111461
- DOI: 10.1371/journal.pone.0020924
The reconstruction of condition-specific transcriptional modules provides new insights in the evolution of yeast AP-1 proteins
Abstract
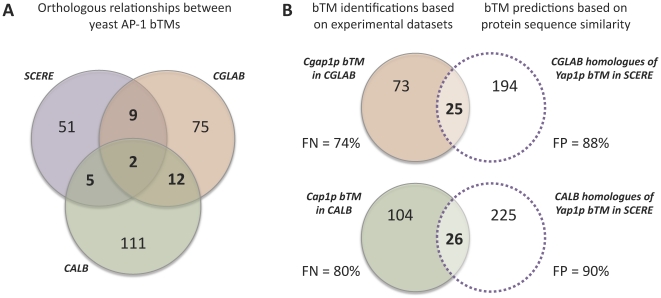
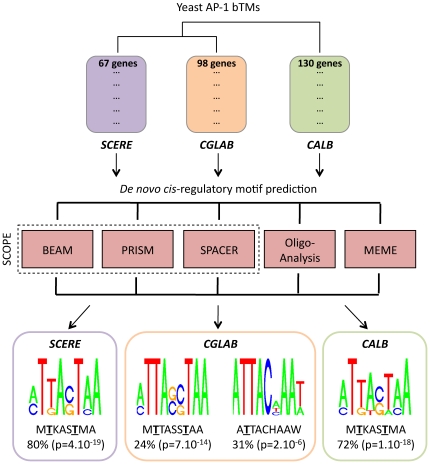
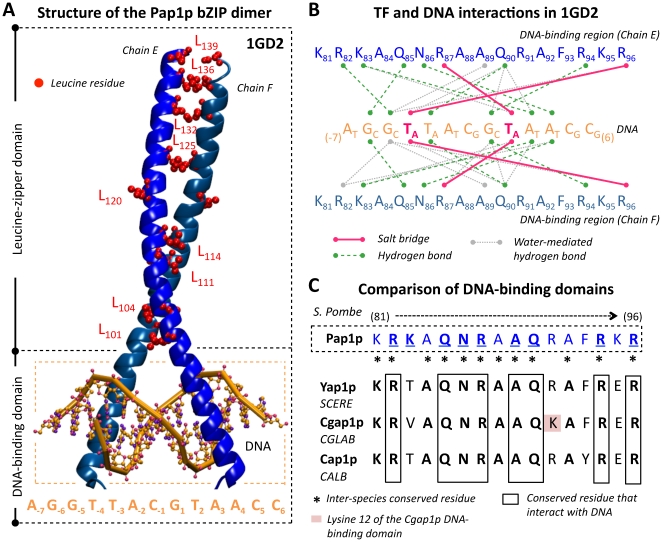
AP-1 proteins are transcription factors (TFs) that belong to the basic leucine zipper family, one of the largest families of TFs in eukaryotic cells. Despite high homology between their DNA binding domains, these proteins are able to recognize diverse DNA motifs. In yeasts, these motifs are referred as YRE (Yap Response Element) and are either seven (YRE-Overlap) or eight (YRE-Adjacent) base pair long. It has been proposed that the AP-1 DNA binding motif preference relies on a single change in the amino acid sequence of the yeast AP-1 TFs (an arginine in the YRE-O binding factors being replaced by a lysine in the YRE-A binding Yaps). We developed a computational approach to infer condition-specific transcriptional modules associated to the orthologous AP-1 protein Yap1p, Cgap1p and Cap1p, in three yeast species: the model yeast Saccharomyces cerevisiae and two pathogenic species Candida glabrata and Candida albicans. Exploitation of these modules in terms of predictions of the protein/DNA regulatory interactions changed our vision of AP-1 protein evolution. Cis-regulatory motif analyses revealed the presence of a conserved adenine in 5' position of the canonical YRE sites. While Yap1p, Cgap1p and Cap1p shared a remarkably low number of target genes, an impressive conservation was observed in the YRE sequences identified by Yap1p and Cap1p. In Candida glabrata, we found that Cgap1p, unlike Yap1p and Cap1p, recognizes YRE-O and YRE-A motifs. These findings were supported by structural data available for the transcription factor Pap1p (Schizosaccharomyces pombe). Thus, whereas arginine and lysine substitutions in Cgap1p and Yap1p proteins were reported as responsible for a specific YRE-O or YRE-A preference, our analyses rather suggest that the ancestral yeast AP-1 protein could recognize both YRE-O and YRE-A motifs and that the arginine/lysine exchange is not the only determinant of the specialization of modern Yaps for one motif or another.
Conflict of interest statement
Figures


Similar articles
-
Analysis of the oxidative stress regulation of the Candida albicans transcription factor, Cap1p.Mol Microbiol. 2000 May;36(3):618-29. doi: 10.1046/j.1365-2958.2000.01877.x. Mol Microbiol. 2000. PMID: 10844651
-
Genome adaptation to chemical stress: clues from comparative transcriptomics in Saccharomyces cerevisiae and Candida glabrata.Genome Biol. 2008;9(11):R164. doi: 10.1186/gb-2008-9-11-r164. Epub 2008 Nov 24. Genome Biol. 2008. PMID: 19025642 Free PMC article.
-
Identification of promoter elements responsible for the regulation of MDR1 from Candida albicans, a major facilitator transporter involved in azole resistance.Microbiology (Reading). 2006 Dec;152(Pt 12):3701-3722. doi: 10.1099/mic.0.29277-0. Microbiology (Reading). 2006. PMID: 17159223
-
Sequence comparison of yeast ATP-binding cassette proteins.Cold Spring Harb Symp Quant Biol. 1995;60:291-307. doi: 10.1101/sqb.1995.060.01.034. Cold Spring Harb Symp Quant Biol. 1995. PMID: 8824403 Review. No abstract available.
-
Redox control of AP-1-like factors in yeast and beyond.Oncogene. 2001 Apr 30;20(19):2336-46. doi: 10.1038/sj.onc.1204384. Oncogene. 2001. PMID: 11402331 Review.
Cited by
-
Yeast AP-1 like transcription factors (Yap) and stress response: a current overview.Microb Cell. 2019 May 28;6(6):267-285. doi: 10.15698/mic2019.06.679. Microb Cell. 2019. PMID: 31172012 Free PMC article. Review.
-
Two residues in the basic region of the yeast transcription factor Yap8 are crucial for its DNA-binding specificity.PLoS One. 2013 Dec 16;8(12):e83328. doi: 10.1371/journal.pone.0083328. eCollection 2013. PLoS One. 2013. PMID: 24358276 Free PMC article.
-
A Network of Paralogous Stress Response Transcription Factors in the Human Pathogen Candida glabrata.Front Microbiol. 2016 May 9;7:645. doi: 10.3389/fmicb.2016.00645. eCollection 2016. Front Microbiol. 2016. PMID: 27242683 Free PMC article.
-
Sequence-specific dynamics of DNA response elements and their flanking sites regulate the recognition by AP-1 transcription factors.Nucleic Acids Res. 2021 Sep 20;49(16):9280-9293. doi: 10.1093/nar/gkab691. Nucleic Acids Res. 2021. PMID: 34387667 Free PMC article.
-
PeAP1-mediated oxidative stress response plays an important role in the growth and pathogenicity of Penicillium expansum.Microbiol Spectr. 2023 Sep 21;11(5):e0380822. doi: 10.1128/spectrum.03808-22. Online ahead of print. Microbiol Spectr. 2023. PMID: 37732795 Free PMC article.
References
-
- Quackenbush J. Computational approaches to analysis of DNA microarray data. Yearb Med Inform. 2006:91–103. - PubMed
-
- Nugent R, Meila M. An overview of clustering applied to molecular biology. Methods Mol Biol. 2010;620:369–404. - PubMed
-
- Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, et al. Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet. 2002;31:255–265. - PubMed
-
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
