

Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase
- PMID: 21690090
- PMCID: PMC3191028
- DOI: 10.1074/jbc.M111.259978
Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase
Abstract
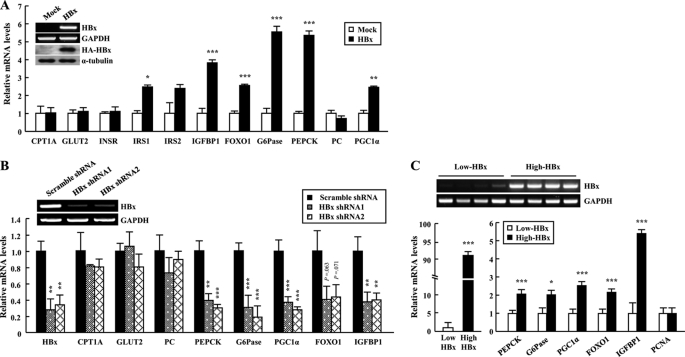
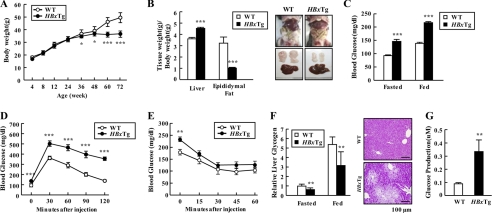
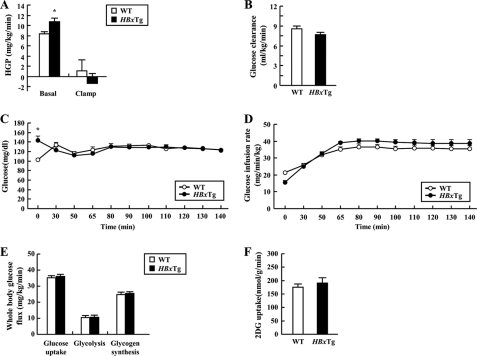
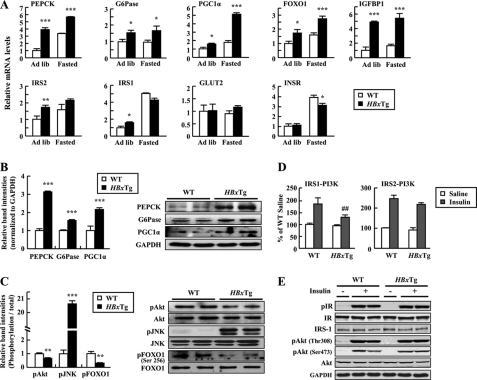
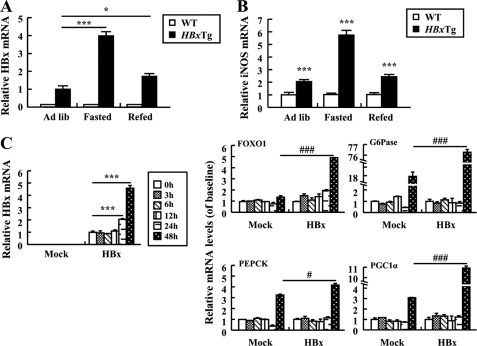
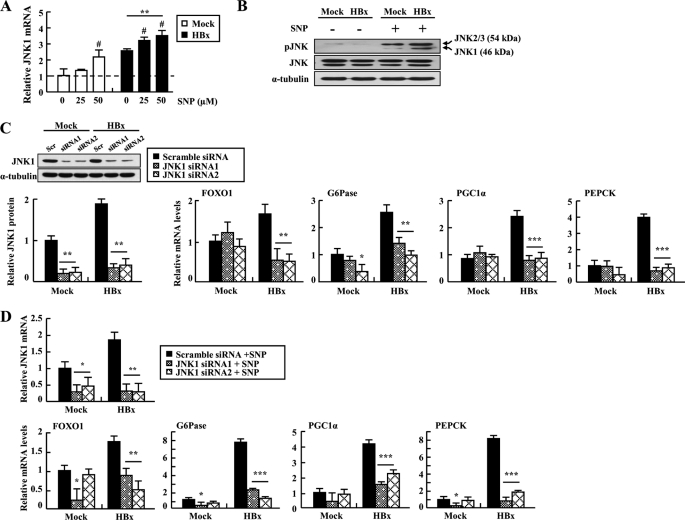
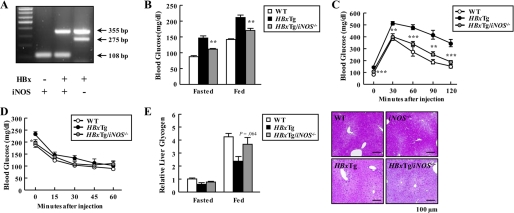
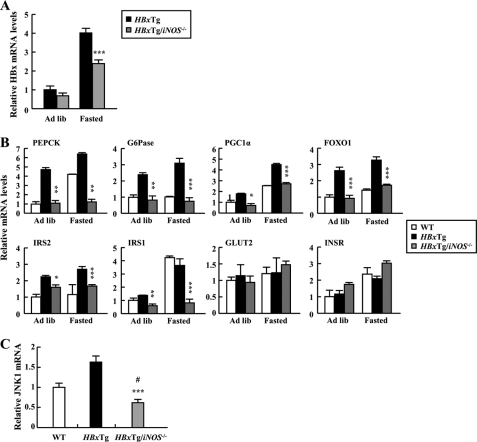
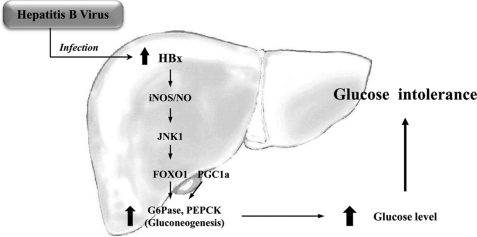
Dysregulation of liver functions leads to insulin resistance causing type 2 diabetes mellitus and is often found in chronic liver diseases. However, the mechanisms of hepatic dysfunction leading to hepatic metabolic disorder are still poorly understood in chronic liver diseases. The current work investigated the role of hepatitis B virus X protein (HBx) in regulating glucose metabolism. We studied HBx-overexpressing (HBxTg) mice and HBxTg mice lacking inducible nitric oxide synthase (iNOS). Here we show that gene expressions of the key gluconeogenic enzymes were significantly increased in HepG2 cells expressing HBx (HepG2-HBx) and in non-tumor liver tissues of hepatitis B virus patients with high levels of HBx expression. In the liver of HBxTg mice, the expressions of gluconeogenic genes were also elevated, leading to hyperglycemia by increasing hepatic glucose production. However, this effect was insufficient to cause systemic insulin resistance. Importantly, the actions of HBx on hepatic glucose metabolism are thought to be mediated via iNOS signaling, as evidenced by the fact that deficiency of iNOS restored HBx-induced hyperglycemia by suppressing the gene expression of gluconeogenic enzymes. Treatment of HepG2-HBx cells with nitric oxide (NO) caused a significant increase in the expression of gluconeogenic genes, but JNK1 inhibition was completely normalized. Furthermore, hyperactivation of JNK1 in the liver of HBxTg mice was also suppressed in the absence of iNOS, indicating the critical role for JNK in the mutual regulation of HBx- and iNOS-mediated glucose metabolism. These findings establish a novel mechanism of HBx-driven hepatic metabolic disorder that is modulated by iNOS-mediated activation of JNK.
Figures
Similar articles
-
Hepatic STAMP2 decreases hepatitis B virus X protein-associated metabolic deregulation.Exp Mol Med. 2012 Oct 31;44(10):622-32. doi: 10.3858/emm.2012.44.10.071. Exp Mol Med. 2012. PMID: 23095254 Free PMC article.
-
Stimulation of inducible nitric oxide by hepatitis B virus transactivator protein HBx requires MTA1 coregulator.J Biol Chem. 2010 Mar 5;285(10):6980-6. doi: 10.1074/jbc.M109.065987. Epub 2009 Dec 18. J Biol Chem. 2010. Retraction in: J Biol Chem. 2017 Mar 17;292(11):4765. doi: 10.1074/jbc.A117.065987 PMID: 20022949 Free PMC article. Retracted.
-
iNOS promotes HBx-induced hepatocellular carcinoma via upregulation of JNK activation.Biochem Biophys Res Commun. 2013 May 31;435(2):244-9. doi: 10.1016/j.bbrc.2013.04.071. Epub 2013 May 3. Biochem Biophys Res Commun. 2013. PMID: 23643810
-
Hepatitis B virus X protein modulates oncogene Yes-associated protein by CREB to promote growth of hepatoma cells.Hepatology. 2012 Dec;56(6):2051-9. doi: 10.1002/hep.25899. Epub 2012 Nov 13. Hepatology. 2012. PMID: 22707013
-
Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma.World J Gastroenterol. 2014 Aug 14;20(30):10238-48. doi: 10.3748/wjg.v20.i30.10238. World J Gastroenterol. 2014. PMID: 25132741 Free PMC article. Review.
Cited by
-
Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences.Int J Mol Sci. 2022 Mar 3;23(5):2787. doi: 10.3390/ijms23052787. Int J Mol Sci. 2022. PMID: 35269929 Free PMC article. Review.
-
A broad investigation of the HBV-mediated changes to primary hepatocyte physiology reveals HBV significantly alters metabolic pathways.Metabolism. 2018 Jun;83:50-59. doi: 10.1016/j.metabol.2018.01.007. Epub 2018 Feb 2. Metabolism. 2018. PMID: 29410347 Free PMC article.
-
Hepatic STAMP2 decreases hepatitis B virus X protein-associated metabolic deregulation.Exp Mol Med. 2012 Oct 31;44(10):622-32. doi: 10.3858/emm.2012.44.10.071. Exp Mol Med. 2012. PMID: 23095254 Free PMC article.
-
Viral hijacking of cellular metabolism.BMC Biol. 2019 Jul 18;17(1):59. doi: 10.1186/s12915-019-0678-9. BMC Biol. 2019. PMID: 31319842 Free PMC article. Review.
-
Transmissible Gastroenteritis Virus Infection Enhances SGLT1 and GLUT2 Expression to Increase Glucose Uptake.PLoS One. 2016 Nov 16;11(11):e0165585. doi: 10.1371/journal.pone.0165585. eCollection 2016. PLoS One. 2016. PMID: 27851758 Free PMC article.
References
-
- Nordlie R. C., Foster J. D., Lange A. J. (1999) Annu. Rev. Nutr. 19, 379–406 - PubMed
-
- Pilkis S. J., Granner D. K. (1992) Annu. Rev. Physiol. 54, 885–909 - PubMed
-
- Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (2003) Diabetes Care 26, Suppl. 1, S5–S20 - PubMed
-
- Kruszynska Y. T., Home P. D., McIntyre N. (1991) Hepatology 14, 103–111 - PubMed
-
- La Vecchia C., Negri E., Decarli A., Franceschi S. (1997) Int. J. Cancer 73, 204–207 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
