

Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis
- PMID: 21596768
- PMCID: PMC3097891
- DOI: 10.1093/brain/awr074
Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis
Abstract
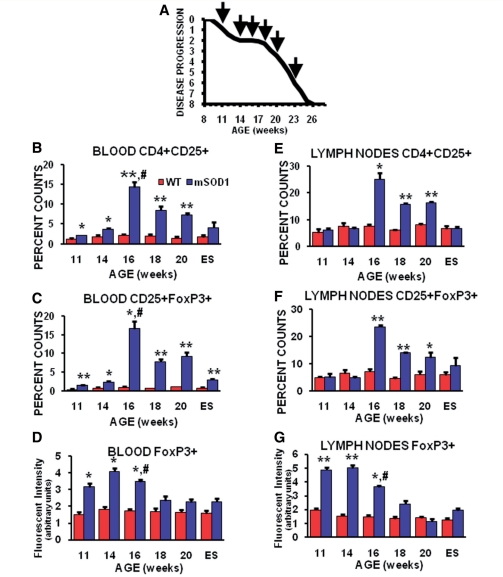
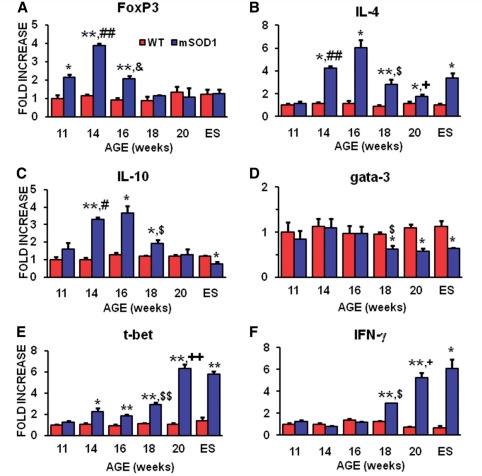
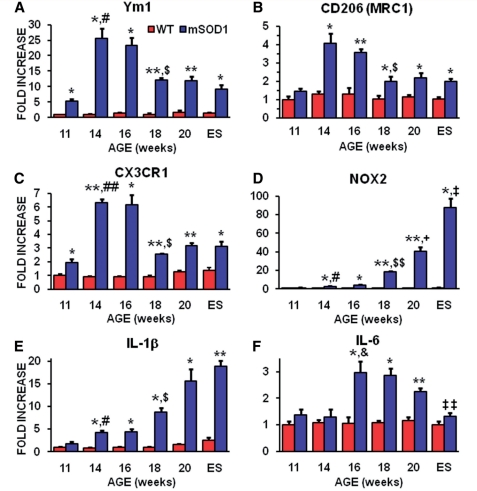
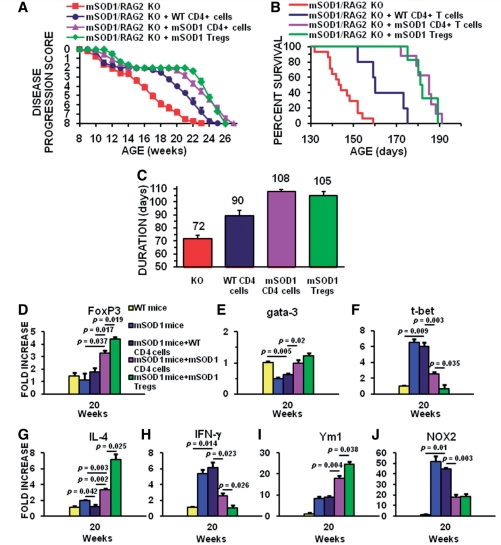
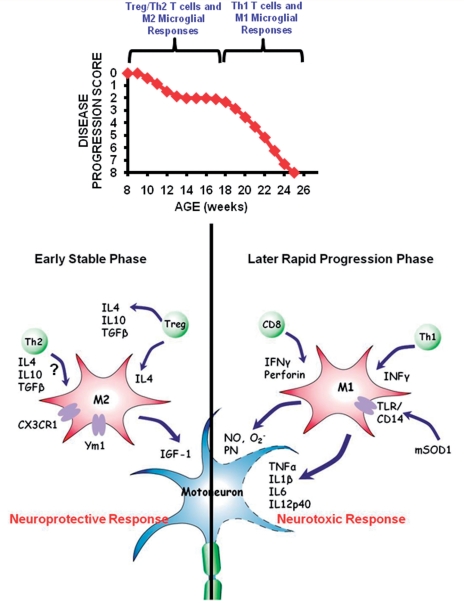
Amyotrophic lateral sclerosis is a relentless and devastating adult-onset neurodegenerative disease with no known cure. In mice with amyotrophic lateral sclerosis, CD4+ T lymphocytes and wild-type microglia potentiate protective inflammatory responses and play a principal role in disease pathoprogression. Using this model, we demonstrate that endogenous T lymphocytes, and more specifically regulatory T lymphocytes, are increased at early slowly progressing stages, augmenting interleukin-4 expression and protective M2 microglia, and are decreased when the disease rapidly accelerates, possibly through the loss of FoxP3 expression in the regulatory T lymphocytes. Without ex vivo activation, the passive transfer of wild-type CD4+ T lymphocytes into amyotrophic lateral sclerosis mice lacking functional T lymphocytes lengthened disease duration and prolonged survival. The passive transfer of endogenous regulatory T lymphocytes from early disease stage mutant Cu2+/Zn2+ superoxide dismutase mice into these amyotrophic lateral sclerosis mice, again without ex vivo activation, were substantially more immunotherapeutic sustaining interleukin-4 levels and M2 microglia, and resulting in lengthened disease duration and prolonged survival; the stable disease phase was extended by 88% using mutant Cu2+/Zn2+ superoxide dismutase regulatory T lymphocytes. A potential mechanism for this enhanced life expectancy may be mediated by the augmented secretion of interleukin-4 from mutant Cu2+/Zn2+ superoxide dismutase regulatory T lymphocytes that directly suppressed the toxic properties of microglia; flow cytometric analyses determined that CD4+/CD25+/FoxP3+ T lymphocytes co-expressed interleukin-4 in the same cell. These observations were extended into the amyotrophic lateral sclerosis patient population where patients with more rapidly progressing disease had decreased numbers of regulatory T lymphocytes; the numbers of regulatory T lymphocytes were inversely correlated with disease progression rates. These data suggest a cellular mechanism whereby endogenous regulatory T lymphocytes are immunocompetent and actively contribute to neuroprotection through their interactions with microglia. Furthermore, these data suggest that immunotherapeutic interventions must begin early in the pathogenic process since immune dysfunction occurs at later stages. Thus, the cumulative mouse and human amyotrophic lateral sclerosis data suggest that increasing the levels of regulatory T lymphocytes in patients with amyotrophic lateral sclerosis at early stages in the disease process may be of therapeutic value, and slow the rate of disease progression and stabilize patients for longer periods of time.
Figures
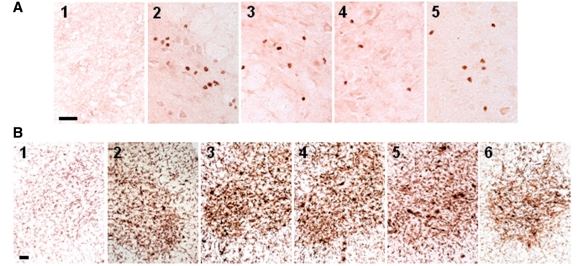
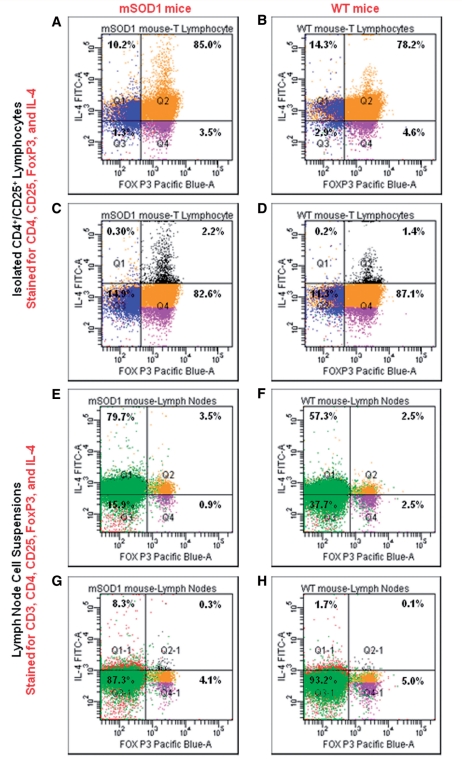
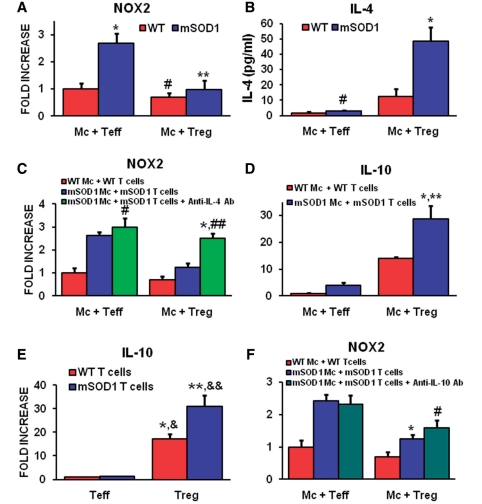
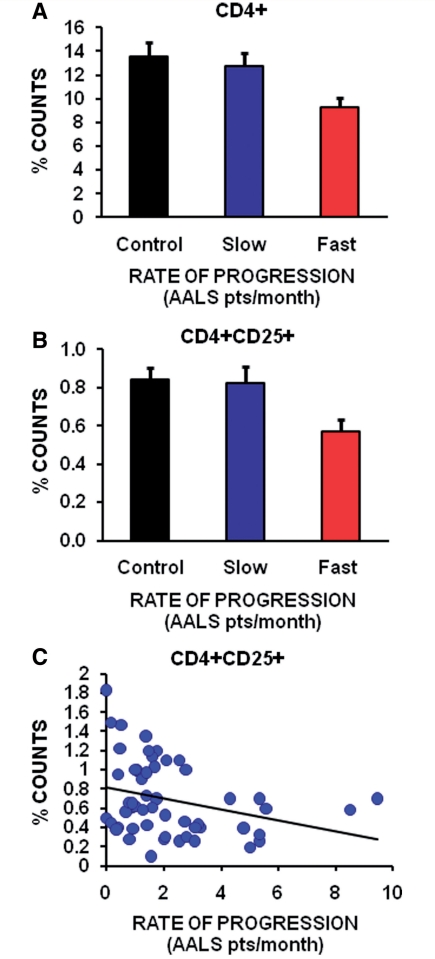
Similar articles
-
Deleterious effects of lymphocytes at the early stage of neurodegeneration in an animal model of amyotrophic lateral sclerosis.J Neuroinflammation. 2011 Feb 23;8:19. doi: 10.1186/1742-2094-8-19. J Neuroinflammation. 2011. PMID: 21345177 Free PMC article.
-
Effect of thymic stimulation of CD4+ T cell expansion on disease onset and progression in mutant SOD1 mice.J Neuroinflammation. 2015 Feb 27;12:40. doi: 10.1186/s12974-015-0254-3. J Neuroinflammation. 2015. PMID: 25889790 Free PMC article.
-
Evaluation of regulatory T lymphocytes and IL2Ra and FOXP3 gene expression in peripheral mononuclear cells from patients with amyotrophic lateral sclerosis.Ir J Med Sci. 2018 Nov;187(4):1065-1071. doi: 10.1007/s11845-018-1793-2. Epub 2018 Mar 25. Ir J Med Sci. 2018. PMID: 29574662
-
Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis.J Neuroimmune Pharmacol. 2013 Sep;8(4):888-99. doi: 10.1007/s11481-013-9489-x. Epub 2013 Jul 25. J Neuroimmune Pharmacol. 2013. PMID: 23881705 Free PMC article. Review.
-
Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis.Neurotherapeutics. 2015 Apr;12(2):364-75. doi: 10.1007/s13311-014-0329-3. Neurotherapeutics. 2015. PMID: 25567201 Free PMC article. Review.
Cited by
-
Friend or foe: the dichotomous impact of T cells on neuro-de/re-generation during aging.Oncotarget. 2017 Jan 24;8(4):7116-7137. doi: 10.18632/oncotarget.12572. Oncotarget. 2017. PMID: 27738345 Free PMC article. Review.
-
T cell responses at diagnosis of amyotrophic lateral sclerosis predict disease progression.Nat Commun. 2022 Nov 8;13(1):6733. doi: 10.1038/s41467-022-34526-9. Nat Commun. 2022. PMID: 36347843 Free PMC article.
-
Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival.EMBO Mol Med. 2013 Jan;5(1):64-79. doi: 10.1002/emmm.201201544. Epub 2012 Nov 9. EMBO Mol Med. 2013. PMID: 23143995 Free PMC article.
-
Effect of complete high-caloric nutrition on the nutritional status and survival rate of amyotrophic lateral sclerosis patients after gastrostomy.Am J Transl Res. 2022 Nov 15;14(11):7842-7851. eCollection 2022. Am J Transl Res. 2022. PMID: 36505314 Free PMC article.
-
Safety of intrathecal injection of Wharton's jelly-derived mesenchymal stem cells in amyotrophic lateral sclerosis therapy.Neural Regen Res. 2019 Feb;14(2):313-318. doi: 10.4103/1673-5374.243723. Neural Regen Res. 2019. PMID: 30531015 Free PMC article.
References
-
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–9. - PubMed
-
- Aloisi F, De Simone R, Columba-Cabezas S, Penna G, Adorini L. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte-macrophage colony-stimulating factor and interaction with Th1 cells. J Immunol. 2000;164:1705–12. - PubMed
-
- Avidan H, Kipnis J, Butovsky O, Caspi RR, Schwartz M. Vaccination with autoantigen protects against aggregated beta-amyloid and glutamate toxicity by controlling microglia: effect of CD4+CD25+ T cells. Eur J Immunol. 2004;34:3434–45. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
