

Protocol matters: which methylome are you actually studying?
- PMID: 21566704
- PMCID: PMC3090160
- DOI: 10.2217/epi.10.36
Protocol matters: which methylome are you actually studying?
Abstract
The field of epigenetics is now capitalizing on the vast number of emerging technologies, largely based on second-generation sequencing, which interrogate DNA methylation status and histone modifications genome-wide. However, getting an exhaustive and unbiased view of a methylome at a reasonable cost is proving to be a significant challenge. In this article, we take a closer look at the impact of the DNA sequence and bias effects introduced to datasets by genome-wide DNA methylation technologies and where possible, explore the bioinformatics tools that deconvolve them. There remains much to be learned about the performance of genome-wide technologies, the data we mine from these assays and how it reflects the actual biology. While there are several methods to interrogate the DNA methylation status genome-wide, our opinion is that no single technique suitably covers the minimum criteria of high coverage and, high resolution at a reasonable cost. In fact, the fraction of the methylome that is studied currently depends entirely on the inherent biases of the protocol employed. There is promise for this to change, as the third generation of sequencing technologies is expected to again 'revolutionize' the way that we study genomes and epigenomes.
Keywords: DNA methylation; epigenetics; high-throughput sequencing; tiling arrays.
Figures
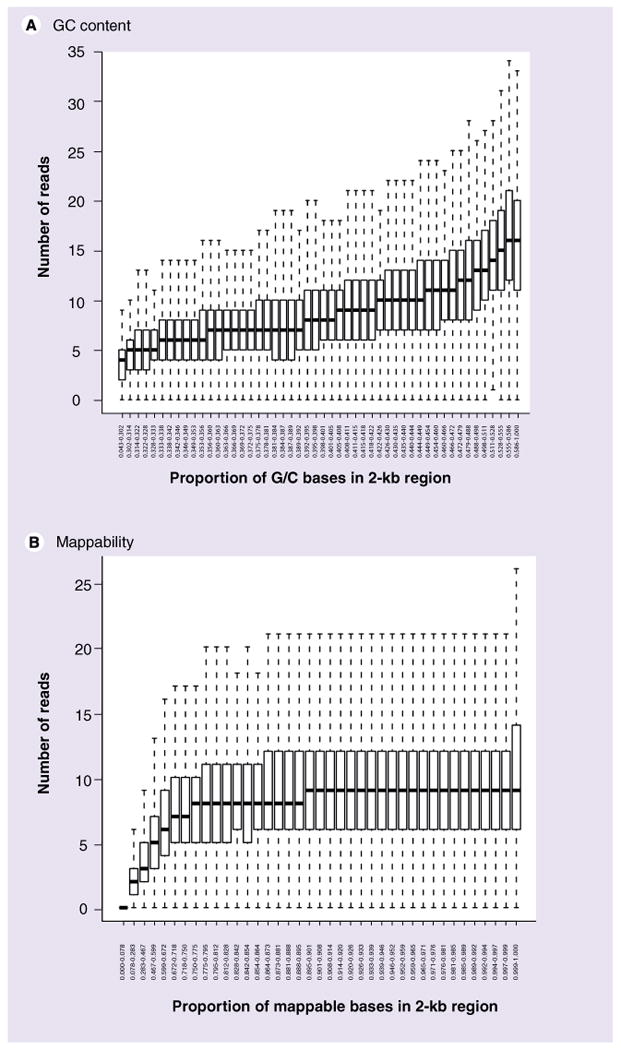
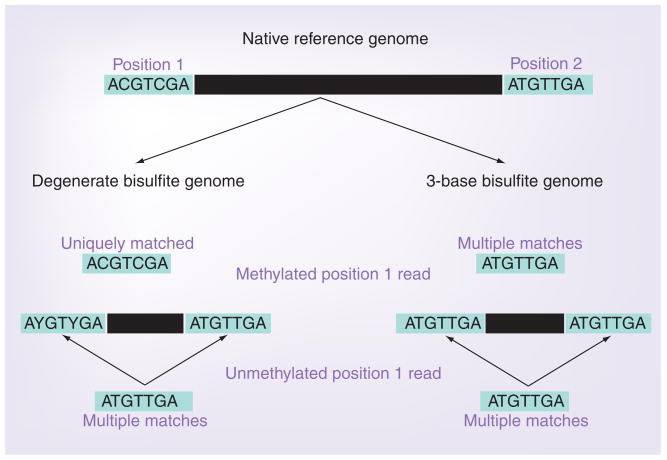
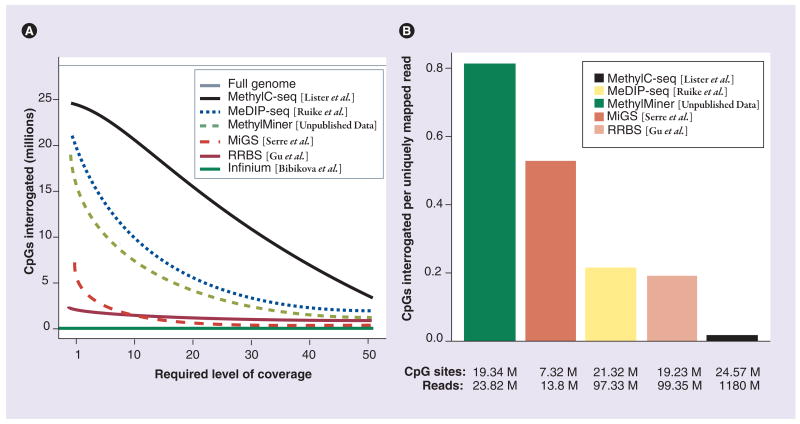
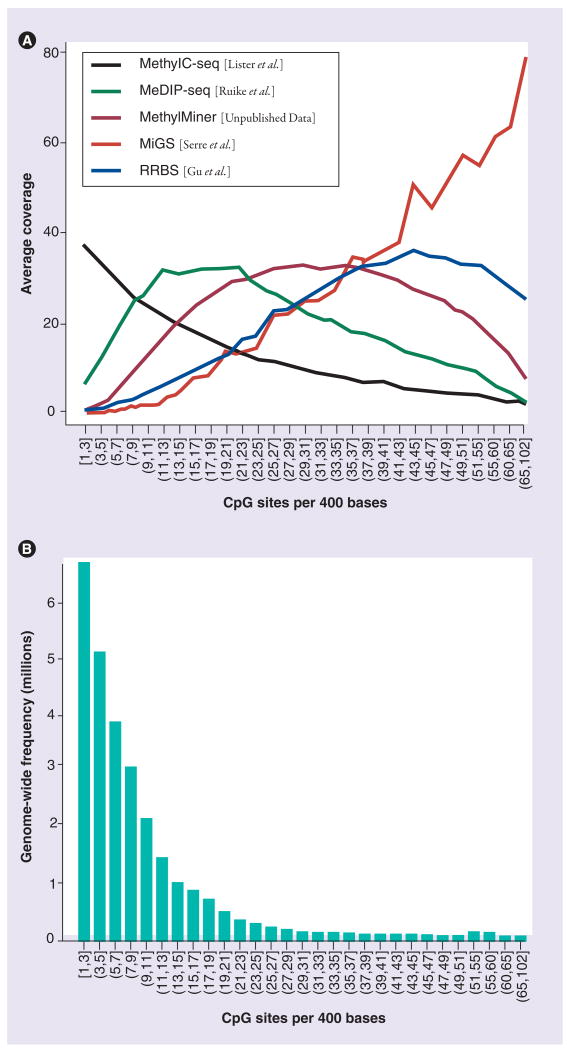
Similar articles
-
Studying the epigenome using next generation sequencing.J Med Genet. 2011 Nov;48(11):721-30. doi: 10.1136/jmedgenet-2011-100242. Epub 2011 Aug 8. J Med Genet. 2011. PMID: 21825079 Review.
-
Nano-MeDIP-seq Methylome Analysis Using Low DNA Concentrations.Methods Mol Biol. 2017;1589:115-138. doi: 10.1007/7651_2015_259. Methods Mol Biol. 2017. PMID: 26138987
-
Genome-scale DNA methylation analysis.Epigenomics. 2010 Feb;2(1):105-17. doi: 10.2217/epi.09.35. Epigenomics. 2010. PMID: 20657796 Free PMC article. Review.
-
Generating Multiple Base-Resolution DNA Methylomes Using Reduced Representation Bisulfite Sequencing.Methods Mol Biol. 2017;1537:279-298. doi: 10.1007/978-1-4939-6685-1_16. Methods Mol Biol. 2017. PMID: 27924600
-
Targeted bisulfite sequencing of the dynamic DNA methylome.Epigenetics Chromatin. 2016 Dec 3;9:55. doi: 10.1186/s13072-016-0105-1. eCollection 2016. Epigenetics Chromatin. 2016. PMID: 27980681 Free PMC article.
Cited by
-
Genome-wide assays that identify and quantify modified cytosines in human disease studies.Epigenetics Chromatin. 2015 Jan 22;8:5. doi: 10.1186/1756-8935-8-5. eCollection 2015. Epigenetics Chromatin. 2015. PMID: 25788985 Free PMC article. Review.
-
Methods for cancer epigenome analysis.Adv Exp Med Biol. 2013;754:313-38. doi: 10.1007/978-1-4419-9967-2_15. Adv Exp Med Biol. 2013. PMID: 22956508 Free PMC article. Review.
-
An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins.Nat Commun. 2014 Dec 12;5:5719. doi: 10.1038/ncomms6719. Nat Commun. 2014. PMID: 25502755 Free PMC article.
-
Recurrent epimutations activate gene body promoters in primary glioblastoma.Genome Res. 2014 May;24(5):761-74. doi: 10.1101/gr.164707.113. Epub 2014 Apr 7. Genome Res. 2014. PMID: 24709822 Free PMC article.
-
Single-CpG resolution mapping of 5-hydroxymethylcytosine by chemical labeling and exonuclease digestion identifies evolutionarily unconserved CpGs as TET targets.Genome Biol. 2016 Mar 29;17:56. doi: 10.1186/s13059-016-0919-y. Genome Biol. 2016. PMID: 27025842 Free PMC article.
References
Bibliography
-
- Clark SJ, Melki J. DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene. 2002;21:5380–5387. - PubMed
-
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. - PubMed
-
- Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nat Genet. 1995;10:20–27. - PubMed
Websites
-
- IUPAC Codes. www.bioinformatics.org/sms/iupac.html.
-
- Sanger Cancer Genome Project. www.sanger.ac.uk/genetics/CGP/
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources