

A founder mutation in the Ashkenazi Jewish population affecting messenger RNA splicing of the CCM2 gene causes cerebral cavernous malformations
- PMID: 21543988
- PMCID: PMC3132303
- DOI: 10.1097/GIM.0b013e318211ff8b
A founder mutation in the Ashkenazi Jewish population affecting messenger RNA splicing of the CCM2 gene causes cerebral cavernous malformations
Abstract
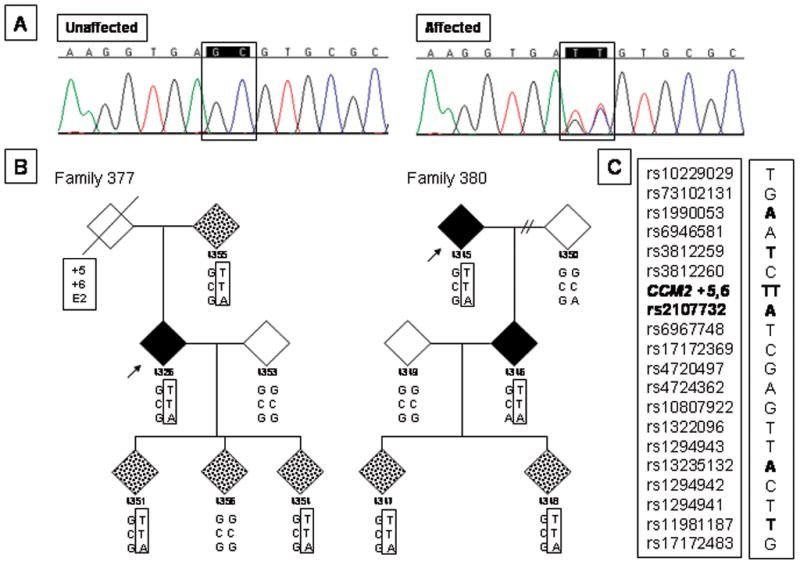
Purpose: Cerebral cavernous malformations can occur sporadically or are caused by mutations in one of three identified genes. Cerebral cavernous malformations often remain clinically silent until a mutation carrier suffers a stroke or seizure. Presymptomatic genetic testing has been valuable to follow and manage cerebral cavernous malformation mutation carriers. During routine diagnostic testing, we identified a two base pair change in seven unrelated people of Ashkenazi Jewish heritage. Because of the location of the variant beyond the invariant splice donor sequence, the change was reported as a variant of unknown significance. In this study, we determined whether this change was a disease-causing mutation and whether it represents a founder mutation in the Ashkenazi Jewish population.
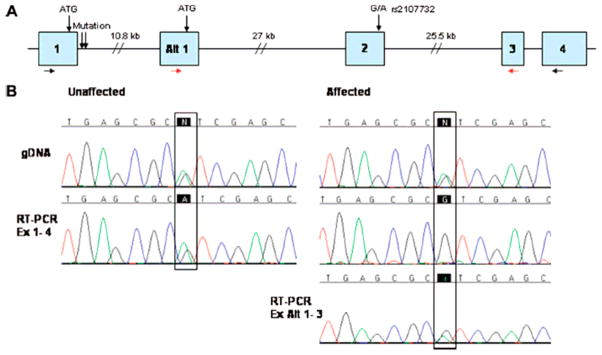
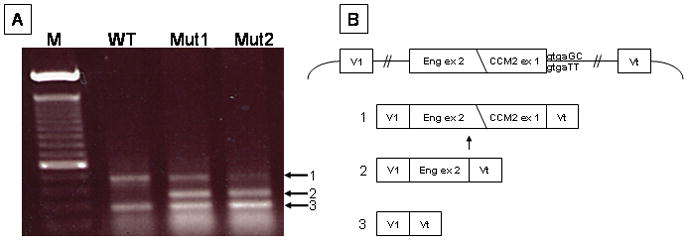
Methods: Transcripts arising from the normal and mutant alleles were examined by reverse transcription-polymerase chain reaction from affected and unaffected Ashkenazi Jewish cerebral cavernous malformation family members. A synthetic splicing system using a chimeric exon was used to visualize the effects of the change on splice donor site utilization.
Results: The two base pair change in CCM2, c.30 + 5_6delinsTT, segregated with affected status in the study families. Reverse transcription-polymerase chain reaction revealed loss of the transcript allele that was in phase with the mutation. The two base pair change, when tested in an in vitro synthetic splicing system, altered splice donor site utilization. Resequencing of the genomic region proximal and distal to the CCM2 gene mutation revealed a common single-nucleotide polymorphism haplotype in affected individuals.
Conclusions: The two base pair change in CCM2, c.30 + 5_6delinsTT, disrupted proper splice donor utilization leading to a degraded transcript. Single nucleotide polymorphism haplotype analysis demonstrated that this mutation was due to a founder in the Ashkenazi Jewish population. These data have the potential to simplify genetic testing for cerebral cavernous malformation in the Ashkenazi Jewish population.
Figures
Similar articles
-
The founder mutation MSH2*1906G-->C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population.Am J Hum Genet. 2002 Dec;71(6):1395-412. doi: 10.1086/345075. Epub 2002 Nov 26. Am J Hum Genet. 2002. PMID: 12454801 Free PMC article.
-
The I1307K APC polymorphism: prevalence in non-Ashkenazi Jews and evidence for a founder effect.Genet Test. 2001 Summer;5(2):141-6. doi: 10.1089/109065701753145628. Genet Test. 2001. PMID: 11551102
-
Identification of a Novel Deletion Mutation (c.1780delG) and a Novel Splice-Site Mutation (c.1412-1G>A) in the CCM1/KRIT1 Gene Associated with Familial Cerebral Cavernous Malformation in the Chinese Population.J Mol Neurosci. 2017 Jan;61(1):8-15. doi: 10.1007/s12031-016-0836-2. Epub 2016 Sep 20. J Mol Neurosci. 2017. PMID: 27649701
-
Cystic fibrosis in Jews: frequency and mutation distribution.Genet Test. 1997;1(1):35-9. doi: 10.1089/gte.1997.1.35. Genet Test. 1997. PMID: 10464623 Review.
-
Explanations for the discrepancy between variant frequency and homozygous disease occurrence: Lessons from Ashkenazi Jewish data.Eur J Med Genet. 2023 Jun;66(6):104765. doi: 10.1016/j.ejmg.2023.104765. Epub 2023 Apr 6. Eur J Med Genet. 2023. PMID: 37028505 Review.
Cited by
-
Genetic genealogy uncovers a founder deletion mutation in the cerebral cavernous malformations 2 gene.Hum Genet. 2022 Nov;141(11):1761-1769. doi: 10.1007/s00439-022-02458-5. Epub 2022 Apr 30. Hum Genet. 2022. PMID: 35488064 Free PMC article.
-
Founder mutations and rare disease in the Arab world.Dis Model Mech. 2024 Jun 1;17(6):dmm050715. doi: 10.1242/dmm.050715. Epub 2024 Jun 26. Dis Model Mech. 2024. PMID: 38922202 Free PMC article. Review.
-
CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions.Neurogenetics. 2013 May;14(2):133-41. doi: 10.1007/s10048-013-0362-0. Epub 2013 Apr 18. Neurogenetics. 2013. PMID: 23595507
-
First large genomic inversion in familial cerebral cavernous malformation identified by whole genome sequencing.Neurogenetics. 2018 Jan;19(1):55-59. doi: 10.1007/s10048-017-0531-7. Epub 2017 Dec 2. Neurogenetics. 2018. PMID: 29197946
-
Cavernous angiomas: deconstructing a neurosurgical disease.J Neurosurg. 2019 Jul 1;131(1):1-13. doi: 10.3171/2019.3.JNS181724. Print 2019 Jul 1. J Neurosurg. 2019. PMID: 31261134 Free PMC article. Review.
References
-
- Laberge-le Couteulx S, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999 Oct;23(2):189–193. - PubMed
-
- Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999 Nov;8(12):2325–2333. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
