

DREME: motif discovery in transcription factor ChIP-seq data
- PMID: 21543442
- PMCID: PMC3106199
- DOI: 10.1093/bioinformatics/btr261
DREME: motif discovery in transcription factor ChIP-seq data
Abstract
Motivation: Transcription factor (TF) ChIP-seq datasets have particular characteristics that provide unique challenges and opportunities for motif discovery. Most existing motif discovery algorithms do not scale well to such large datasets, or fail to report many motifs associated with cofactors of the ChIP-ed TF.
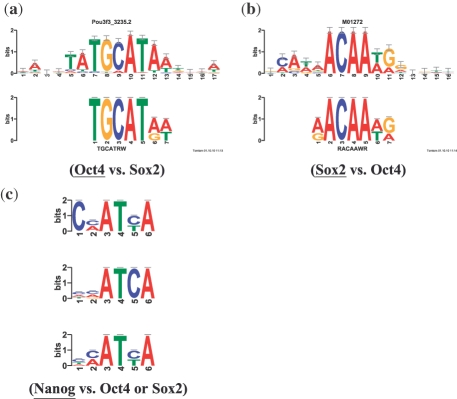
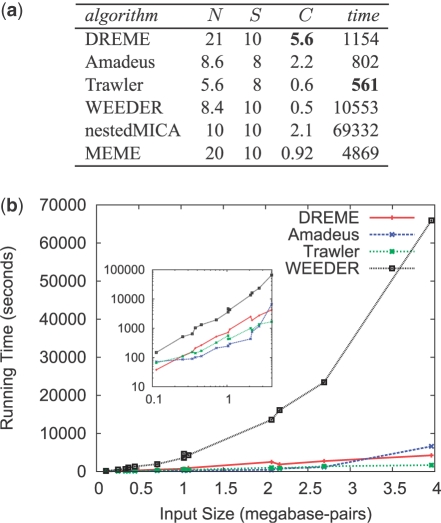
Results: We present DREME, a motif discovery algorithm specifically designed to find the short, core DNA-binding motifs of eukaryotic TFs, and optimized to analyze very large ChIP-seq datasets in minutes. Using DREME, we discover the binding motifs of the the ChIP-ed TF and many cofactors in mouse ES cell (mESC), mouse erythrocyte and human cell line ChIP-seq datasets. For example, in mESC ChIP-seq data for the TF Esrrb, we discover the binding motifs for eight cofactor TFs important in the maintenance of pluripotency. Several other commonly used algorithms find at most two cofactor motifs in this same dataset. DREME can also perform discriminative motif discovery, and we use this feature to provide evidence that Sox2 and Oct4 do not bind in mES cells as an obligate heterodimer. DREME is much faster than many commonly used algorithms, scales linearly in dataset size, finds multiple, non-redundant motifs and reports a reliable measure of statistical significance for each motif found. DREME is available as part of the MEME Suite of motif-based sequence analysis tools (http://meme.nbcr.net).
Figures

Similar articles
-
MEME-ChIP: motif analysis of large DNA datasets.Bioinformatics. 2011 Jun 15;27(12):1696-7. doi: 10.1093/bioinformatics/btr189. Epub 2011 Apr 12. Bioinformatics. 2011. PMID: 21486936 Free PMC article.
-
EXTREME: an online EM algorithm for motif discovery.Bioinformatics. 2014 Jun 15;30(12):1667-73. doi: 10.1093/bioinformatics/btu093. Epub 2014 Feb 14. Bioinformatics. 2014. PMID: 24532725 Free PMC article.
-
A Fast Cluster Motif Finding Algorithm for ChIP-Seq Data Sets.Biomed Res Int. 2015;2015:218068. doi: 10.1155/2015/218068. Epub 2015 Jul 5. Biomed Res Int. 2015. PMID: 26236718 Free PMC article.
-
An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data.Brief Bioinform. 2018 Sep 28;19(5):1069-1081. doi: 10.1093/bib/bbx026. Brief Bioinform. 2018. PMID: 28334268 Review.
-
Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond.Cell Cycle. 2014;13(18):2847-52. doi: 10.4161/15384101.2014.949201. Cell Cycle. 2014. PMID: 25486472 Free PMC article. Review.
Cited by
-
PreDREM: a database of predicted DNA regulatory motifs from 349 human cell and tissue samples.Database (Oxford). 2015 Feb 27;2015:bav007. doi: 10.1093/database/bav007. Print 2015. Database (Oxford). 2015. PMID: 25725063 Free PMC article.
-
HEPeak: an HMM-based exome peak-finding package for RNA epigenome sequencing data.BMC Genomics. 2015;16 Suppl 4(Suppl 4):S2. doi: 10.1186/1471-2164-16-S4-S2. Epub 2015 Apr 21. BMC Genomics. 2015. PMID: 25917296 Free PMC article.
-
Nanopore long-read RNA-seq and absolute quantification delineate transcription dynamics in early embryo development of an insect pest.Sci Rep. 2021 Apr 12;11(1):7878. doi: 10.1038/s41598-021-86753-7. Sci Rep. 2021. PMID: 33846393 Free PMC article.
-
Photosynthetic Genes and Genes Associated with the C4 Trait in Maize Are Characterized by a Unique Class of Highly Regulated Histone Acetylation Peaks on Upstream Promoters.Plant Physiol. 2015 Aug;168(4):1378-88. doi: 10.1104/pp.15.00934. Epub 2015 Jun 25. Plant Physiol. 2015. PMID: 26111542 Free PMC article.
-
The RNA binding protein FgRbp1 regulates specific pre-mRNA splicing via interacting with U2AF23 in Fusarium.Nat Commun. 2021 May 11;12(1):2661. doi: 10.1038/s41467-021-22917-3. Nat Commun. 2021. PMID: 33976182 Free PMC article.
References
-
- Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995;3:21–29. - PubMed
-
- Barash Y, et al. A simple hyper-geometric approach for discovering putative transcription factor binding sites. In: Gascuel O, Moret BME, editors. Algorithms in Bioinformatics: Proceedings of the First International Workshop. 2001. Vol. 2149 in Lecture Notes in Computer Science, Springer, pp. 278–293.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
