

Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection
- PMID: 21518788
- PMCID: PMC3191985
- DOI: 10.1128/IAI.00067-11
Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection
Erratum in
- Infect Immun. 2013 Nov;81(11):4323
Abstract
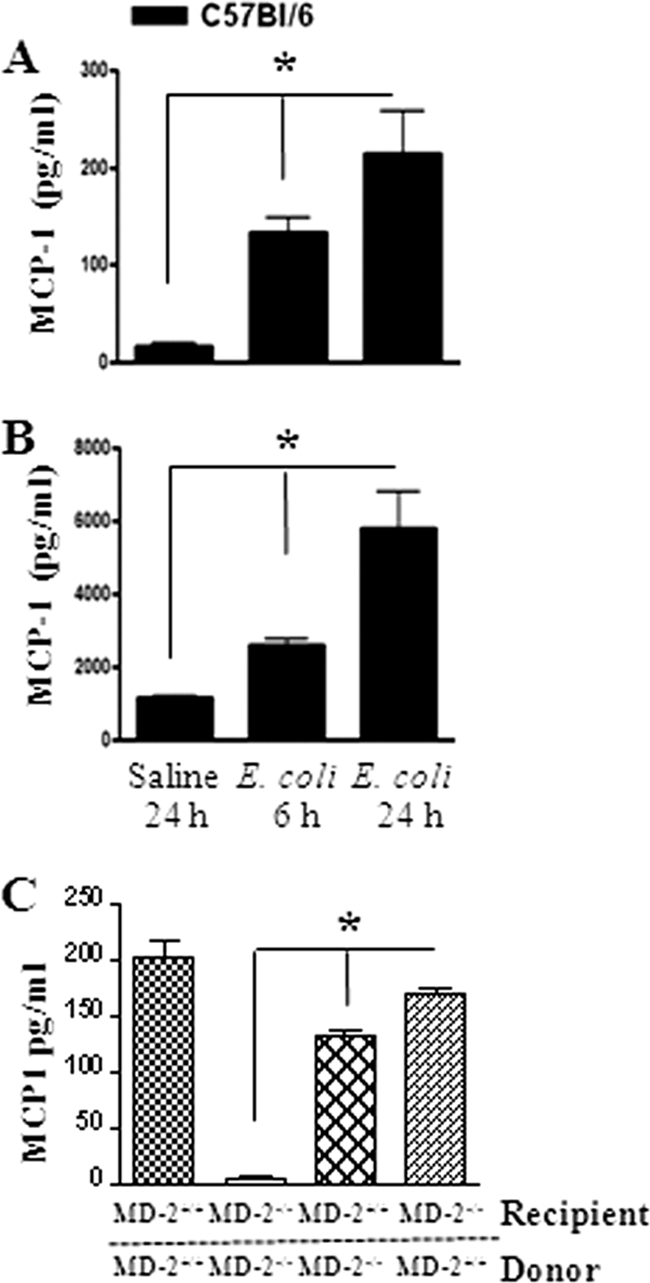
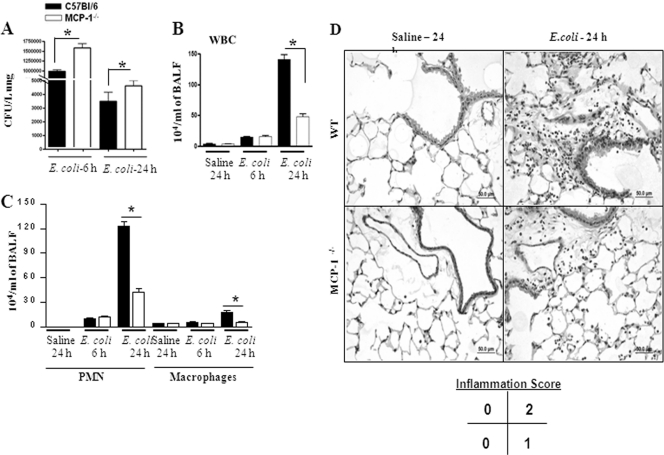
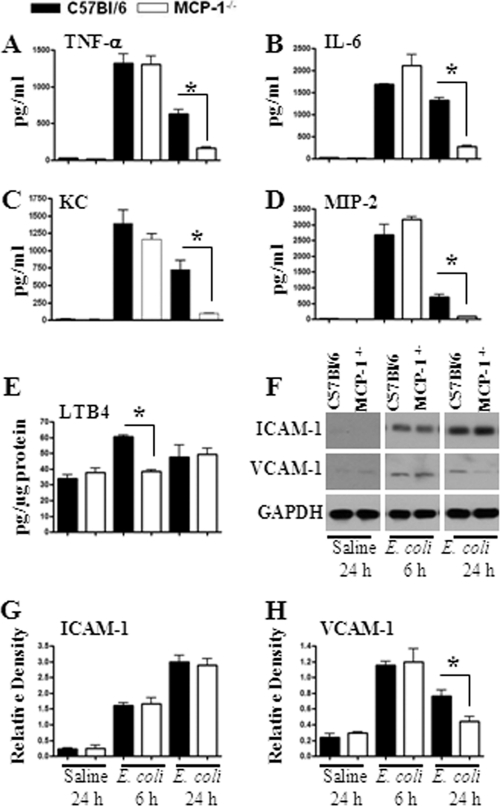
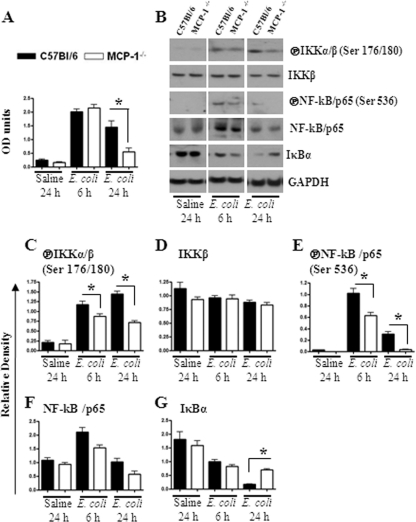
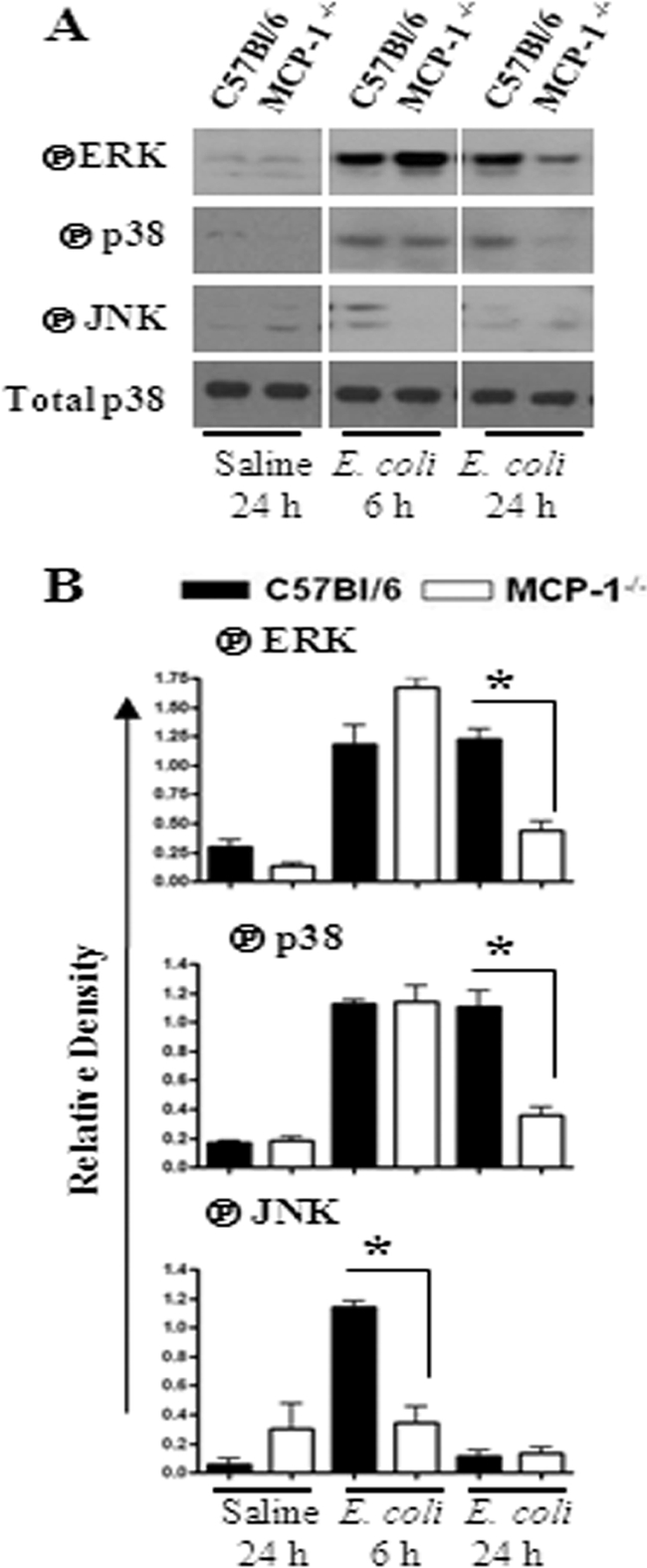
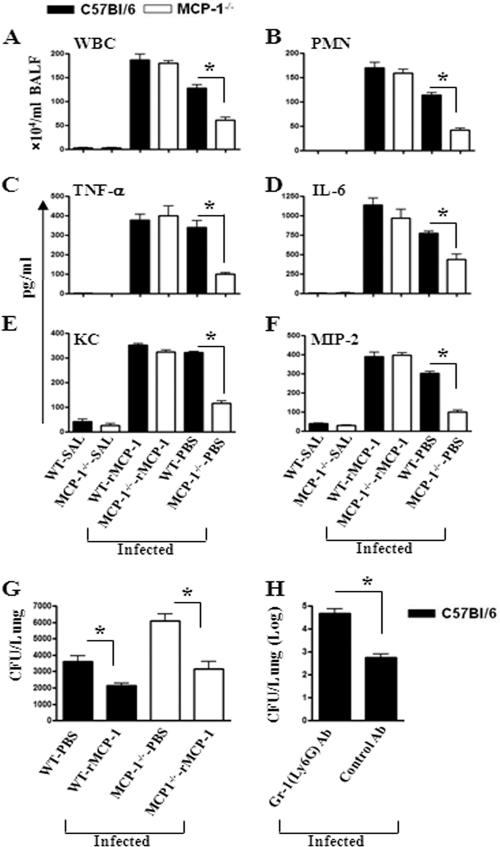
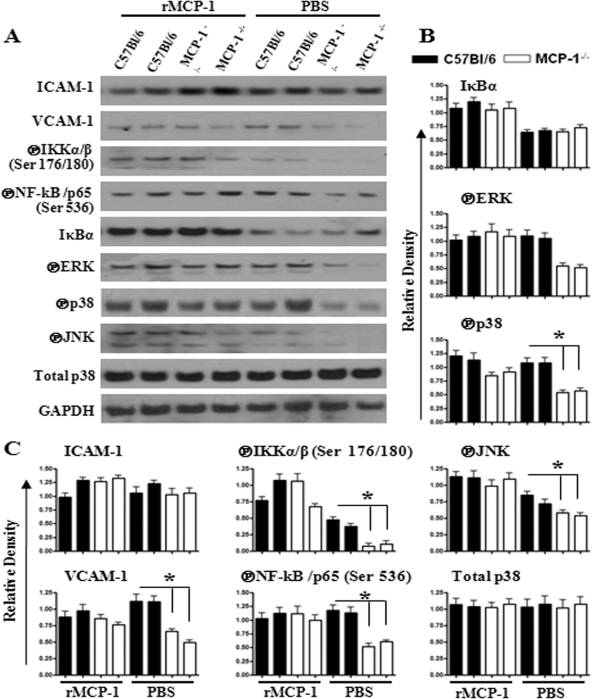
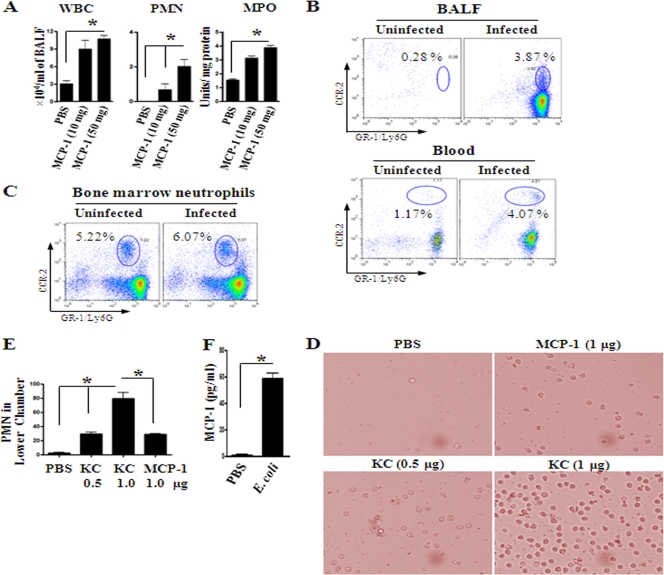
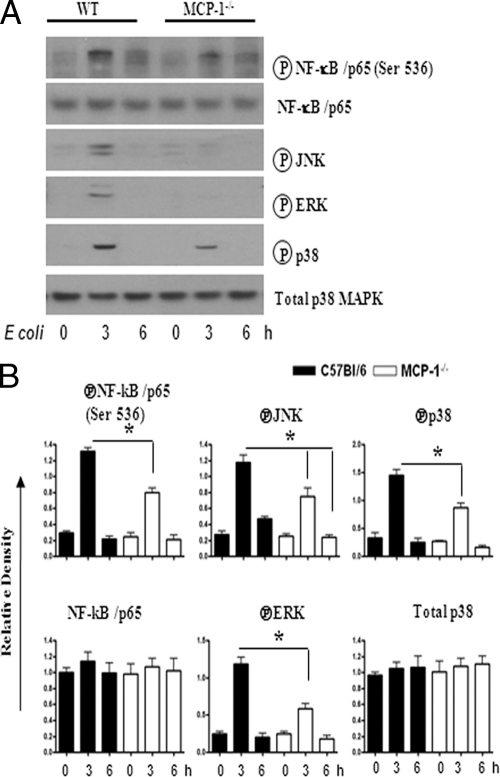
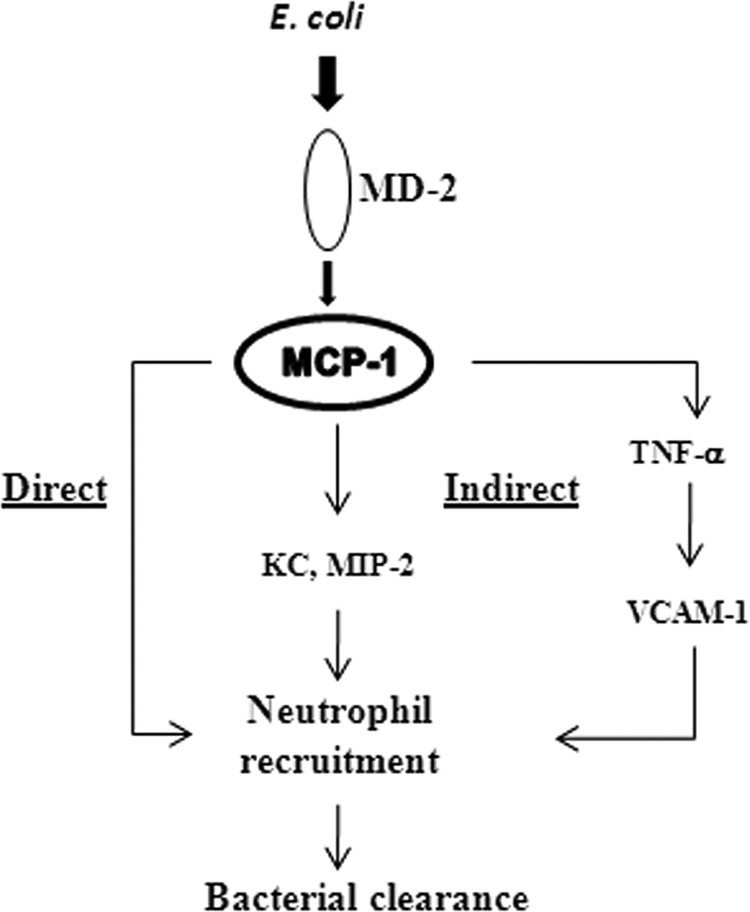
Neutrophil accumulation is a critical event to clear bacteria. Since uncontrolled neutrophil recruitment can cause severe lung damage, understanding neutrophil trafficking mechanisms is important to attenuate neutrophil-mediated damage. While monocyte chemoattractant protein 1 (MCP-1) is known to be a monocyte chemoattractant, its role in pulmonary neutrophil-mediated host defense against Gram-negative bacterial infection is not understood. We hypothesized that MCP-1/chemokine (C-C motif) ligand 2 is important for neutrophil-mediated host defense. Reduced bacterial clearance in the lungs was observed in MCP-1(-/-) mice following Escherichia coli infection. Neutrophil influx, along with cytokines/chemokines, leukotriene B(4) (LTB(4)), and vascular cell adhesion molecule 1 levels in the lungs, was reduced in MCP-1(-/-) mice after infection. E. coli-induced activation of NF-κB and mitogen-activated protein kinases in the lung was also reduced in MCP-1(-/-) mice. Administration of intratracheal recombinant MCP-1 (rMCP-1) to MCP-1(-/-) mice induced pulmonary neutrophil influx and cytokine/chemokine responses in the presence or absence of E. coli infection. Our in vitro migration experiment demonstrates MCP-1-mediated neutrophil chemotaxis. Notably, chemokine receptor 2 is expressed on lung and blood neutrophils, which are increased upon E. coli infection. Furthermore, our findings show that neutrophil depletion impairs E. coli clearance and that exogenous rMCP-1 after infection improves bacterial clearance in the lungs. Overall, these new findings demonstrate that E. coli-induced MCP-1 causes neutrophil recruitment directly via chemotaxis as well as indirectly via modulation of keratinocyte cell-derived chemokine, macrophage inflammatory protein 2, and LTB(4).
Figures
Similar articles
-
Intrapulmonary G-CSF rescues neutrophil recruitment to the lung and neutrophil release to blood in Gram-negative bacterial infection in MCP-1-/- mice.J Immunol. 2012 Dec 15;189(12):5849-59. doi: 10.4049/jimmunol.1200585. Epub 2012 Nov 5. J Immunol. 2012. PMID: 23129755 Free PMC article.
-
Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia.J Immunol. 2007 Mar 1;178(5):3153-60. doi: 10.4049/jimmunol.178.5.3153. J Immunol. 2007. PMID: 17312163
-
NOD2 signaling contributes to host defense in the lungs against Escherichia coli infection.Infect Immun. 2012 Jul;80(7):2558-69. doi: 10.1128/IAI.06230-11. Epub 2012 Apr 30. Infect Immun. 2012. Retraction in: Infect Immun. 2015 May;83(5):2199. doi: 10.1128/IAI.00200-15. PMID: 22547547 Free PMC article. Retracted.
-
MCP-1: Function, regulation, and involvement in disease.Int Immunopharmacol. 2021 Dec;101(Pt B):107598. doi: 10.1016/j.intimp.2021.107598. Epub 2021 May 20. Int Immunopharmacol. 2021. PMID: 34233864 Free PMC article. Review.
-
New Pharmacological Tools to Target Leukocyte Trafficking in Lung Disease.Front Immunol. 2021 Jul 21;12:704173. doi: 10.3389/fimmu.2021.704173. eCollection 2021. Front Immunol. 2021. PMID: 34367163 Free PMC article. Review.
Cited by
-
NO-dependent attenuation of TPA-induced immunoinflammatory skin changes in Balb/c mice by pindolol, heptaminol or ATRA, but not by verapamil.Oncotarget. 2016 Jul 26;7(30):47576-47585. doi: 10.18632/oncotarget.10217. Oncotarget. 2016. PMID: 27374093 Free PMC article.
-
Cellular Immune Response against Nontypeable Haemophilus influenzae Infecting the Preinflamed Middle Ear of the Junbo Mouse.Infect Immun. 2019 Nov 18;87(12):e00689-19. doi: 10.1128/IAI.00689-19. Print 2019 Dec. Infect Immun. 2019. PMID: 31548315 Free PMC article.
-
MyD88 signaling regulates both host defense and immunopathogenesis during pneumocystis infection.J Immunol. 2014 Jan 1;192(1):282-92. doi: 10.4049/jimmunol.1301431. Epub 2013 Nov 29. J Immunol. 2014. PMID: 24293628 Free PMC article.
-
A Dual Role for the Plasminogen Activator Protease During the Preinflammatory Phase of Primary Pneumonic Plague.J Infect Dis. 2020 Jul 6;222(3):407-416. doi: 10.1093/infdis/jiaa094. J Infect Dis. 2020. PMID: 32128567 Free PMC article.
-
Pulmonary Expression of Interleukin-17 Contributes to Neutrophil Infiltration into the Lungs during Pneumonic Plague.Infect Immun. 2023 Jul 18;91(7):e0013123. doi: 10.1128/iai.00131-23. Epub 2023 Jun 20. Infect Immun. 2023. PMID: 37338372 Free PMC article.
References
-
- Abraham E. 2003. Neutrophils and acute lung injury. Crit. Care Med. 31:S195–S199 - PubMed
-
- Amano H., et al. 2004. Essential contribution of monocyte chemoattractant protein-1/C-C chemokine ligand-2 to resolution and repair processes in acute bacterial pneumonia. J. Immunol. 172:398–409 - PubMed
-
- Bazan-Socha S., Bukiej A., Marcinkiewicz C., Musial J. 2005. Integrins in pulmonary inflammatory diseases. Curr. Pharm. Des. 11:893–901 - PubMed
-
- Bhatia M., et al. 2008. Treatment with bindarit, an inhibitor of MCP-1 synthesis, protects mice against trinitrobenzene sulfonic acid-induced colitis. Inflamm. Res. 57:464–471 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
