

Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research
- PMID: 21473983
- PMCID: PMC3071919
- DOI: 10.1016/j.ajhg.2011.03.013
Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research
Abstract
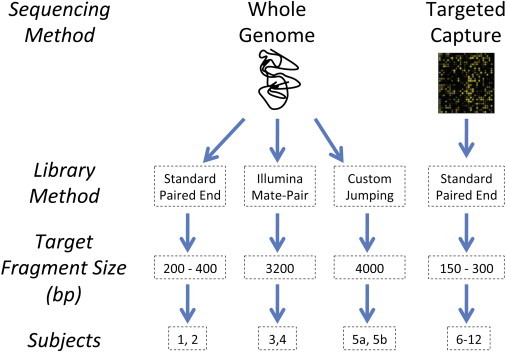
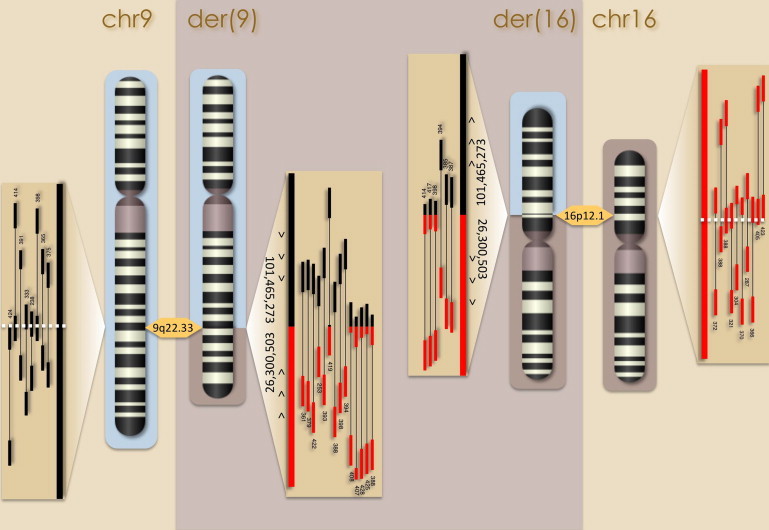
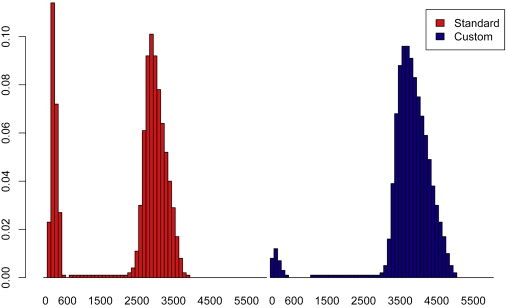
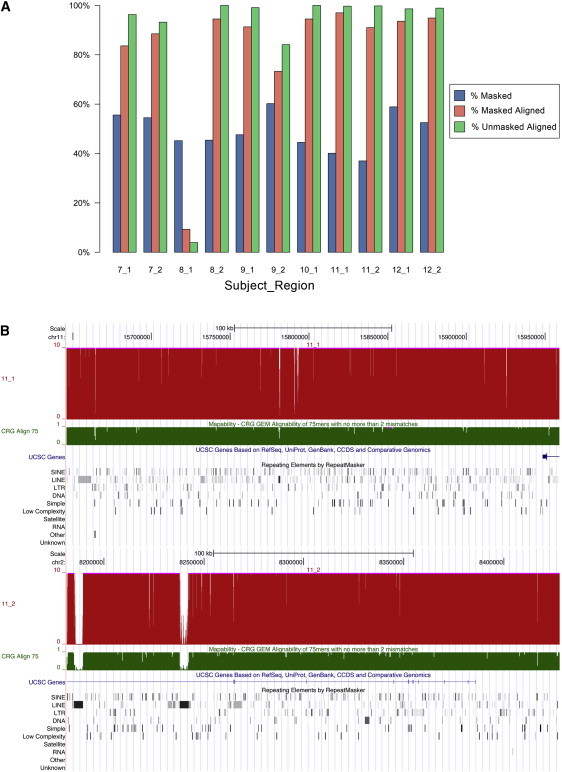
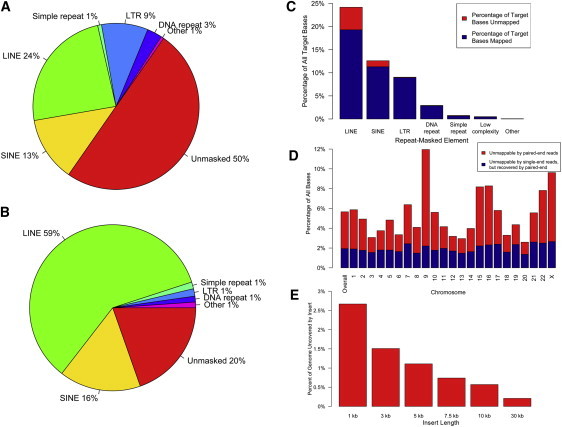
The contribution of balanced chromosomal rearrangements to complex disorders remains unclear because they are not detected routinely by genome-wide microarrays and clinical localization is imprecise. Failure to consider these events bypasses a potentially powerful complement to single nucleotide polymorphism and copy-number association approaches to complex disorders, where much of the heritability remains unexplained. To capitalize on this genetic resource, we have applied optimized sequencing and analysis strategies to test whether these potentially high-impact variants can be mapped at reasonable cost and throughput. By using a whole-genome multiplexing strategy, rearrangement breakpoints could be delineated at a fraction of the cost of standard sequencing. For rearrangements already mapped regionally by karyotyping and fluorescence in situ hybridization, a targeted approach enabled capture and sequencing of multiple breakpoints simultaneously. Importantly, this strategy permitted capture and unique alignment of up to 97% of repeat-masked sequences in the targeted regions. Genome-wide analyses estimate that only 3.7% of bases should be routinely omitted from genomic DNA capture experiments. Illustrating the power of these approaches, the rearrangement breakpoints were rapidly defined to base pair resolution and revealed unexpected sequence complexity, such as co-occurrence of inversion and translocation as an underlying feature of karyotypically balanced alterations. These findings have implications ranging from genome annotation to de novo assemblies and could enable sequencing screens for structural variations at a cost comparable to that of microarrays in standard clinical practice.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Deciphering the complexity of simple chromosomal insertions by genome sequencing.Hum Genet. 2021 Feb;140(2):361-380. doi: 10.1007/s00439-020-02210-x. Epub 2020 Jul 29. Hum Genet. 2021. PMID: 32728808
-
Additional cryptic CNVs in mentally retarded patients with apparently balanced karyotypes.Eur J Med Genet. 2010 Sep-Oct;53(5):227-33. doi: 10.1016/j.ejmg.2010.06.003. Epub 2010 Jun 11. Eur J Med Genet. 2010. PMID: 20542150
-
The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes.J Med Genet. 2005 Jan;42(1):8-16. doi: 10.1136/jmg.2004.024141. J Med Genet. 2005. PMID: 15635069 Free PMC article.
-
Characterising chromosome rearrangements: recent technical advances in molecular cytogenetics.Heredity (Edinb). 2012 Jan;108(1):75-85. doi: 10.1038/hdy.2011.100. Epub 2011 Nov 16. Heredity (Edinb). 2012. PMID: 22086080 Free PMC article. Review.
-
Cellular and genomic approaches for exploring structural chromosomal rearrangements.Chromosome Res. 2020 Mar;28(1):19-30. doi: 10.1007/s10577-020-09626-1. Epub 2020 Jan 13. Chromosome Res. 2020. PMID: 31933061 Free PMC article. Review.
Cited by
-
Evaluation of four genetic variants in han chinese subjects with high myopia.J Ophthalmol. 2015;2015:729463. doi: 10.1155/2015/729463. Epub 2015 Jan 5. J Ophthalmol. 2015. PMID: 25628894 Free PMC article.
-
Unbalanced translocations arise from diverse mutational mechanisms including chromothripsis.Genome Res. 2015 Jul;25(7):937-47. doi: 10.1101/gr.191247.115. Epub 2015 Jun 12. Genome Res. 2015. PMID: 26070663 Free PMC article.
-
Deciphering the pathogenic consequences of chromosomal aberrations in human genetic disease.Mol Cytogenet. 2014 Dec 19;7(1):100. doi: 10.1186/s13039-014-0100-9. eCollection 2014. Mol Cytogenet. 2014. PMID: 25606056 Free PMC article.
-
A Balanced Translocation in Kallmann Syndrome Implicates a Long Noncoding RNA, RMST, as a GnRH Neuronal Regulator.J Clin Endocrinol Metab. 2020 Mar 1;105(3):e231-44. doi: 10.1210/clinem/dgz011. J Clin Endocrinol Metab. 2020. PMID: 31628846 Free PMC article.
-
Comprehensive clinically oriented workflow for nucleotide level resolution and interpretation in prenatal diagnosis of de novo apparently balanced chromosomal translocations in their genomic landscape.Hum Genet. 2020 Apr;139(4):531-543. doi: 10.1007/s00439-020-02121-x. Epub 2020 Feb 6. Hum Genet. 2020. PMID: 32030560 Free PMC article.
References
-
- Rowley J.D. Chromosome abnormalities in leukemia. Haematol. Blood Transfus. 1979;23:43–52. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
