

Probing the determinants of diacylglycerol binding affinity in the C1B domain of protein kinase Cα
- PMID: 21419781
- PMCID: PMC3489171
- DOI: 10.1016/j.jmb.2011.03.020
Probing the determinants of diacylglycerol binding affinity in the C1B domain of protein kinase Cα
Abstract
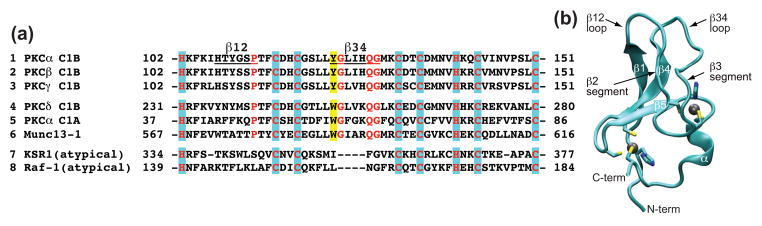
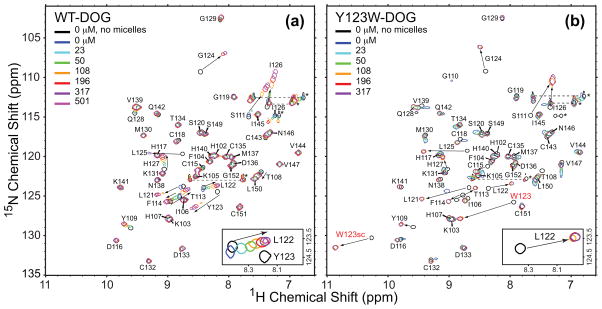
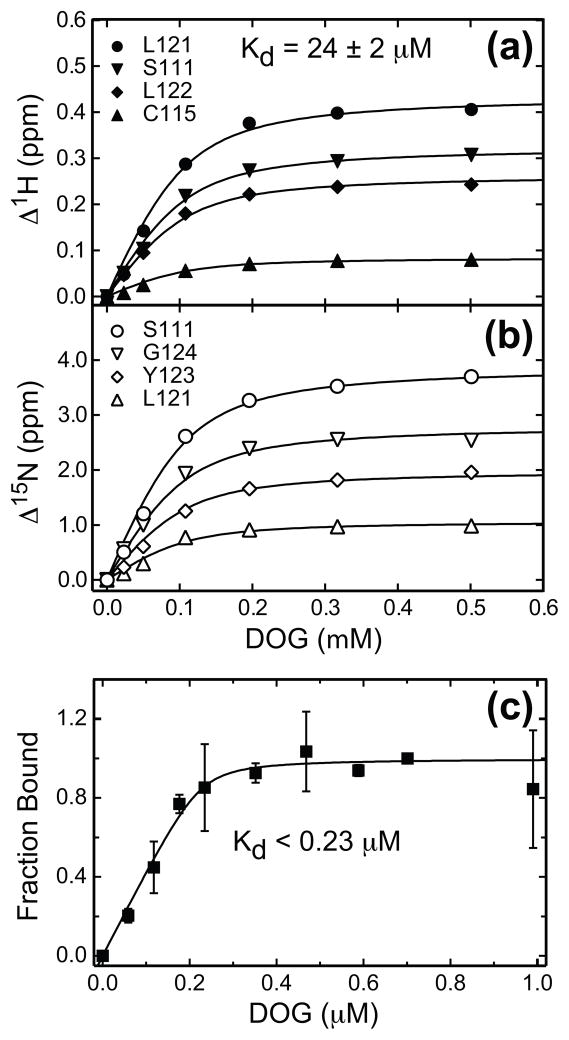
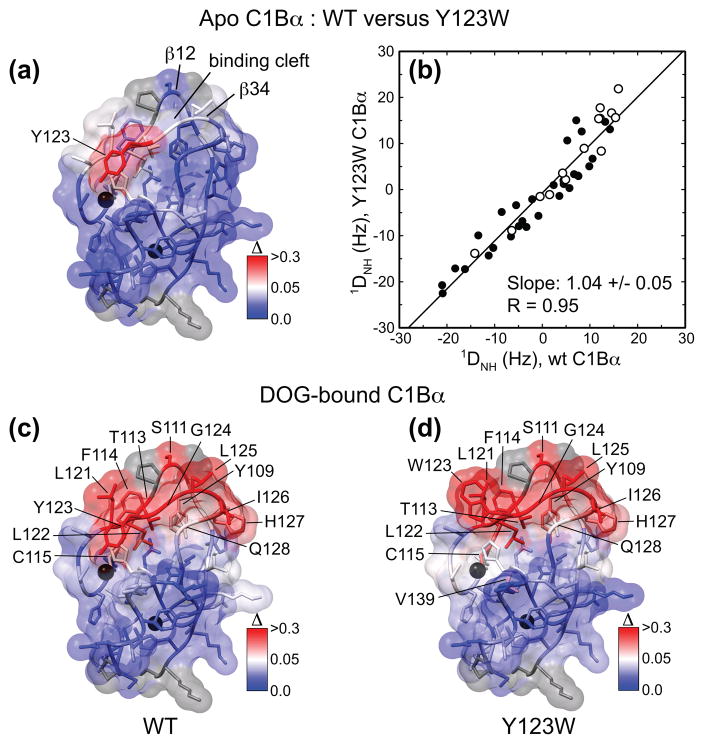
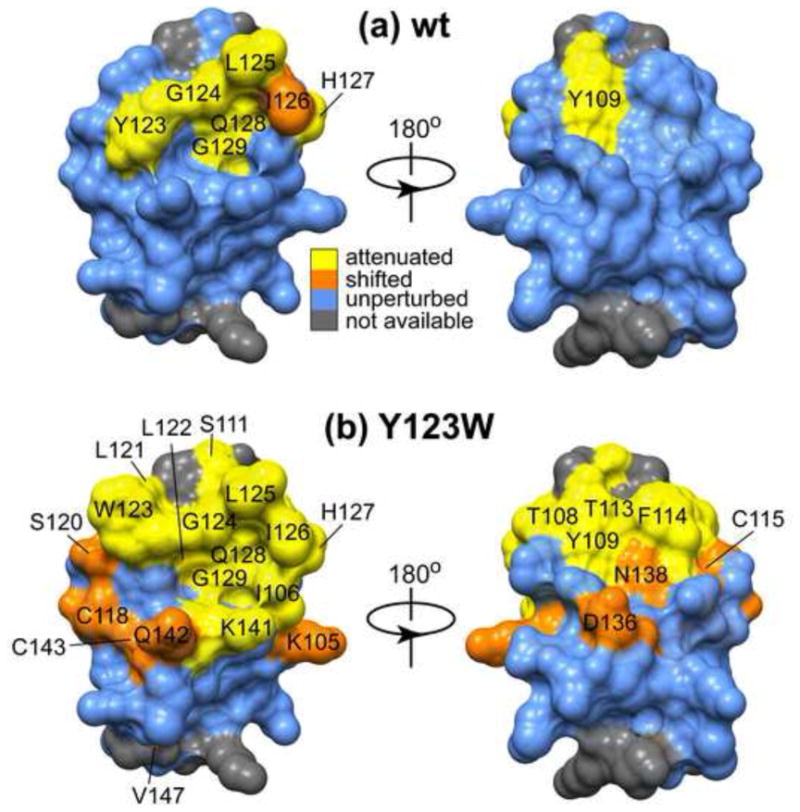
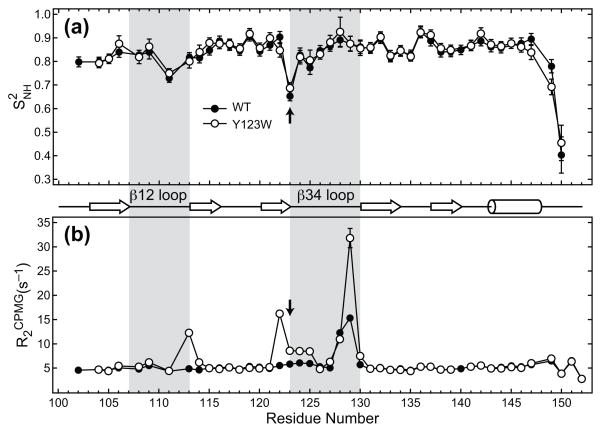
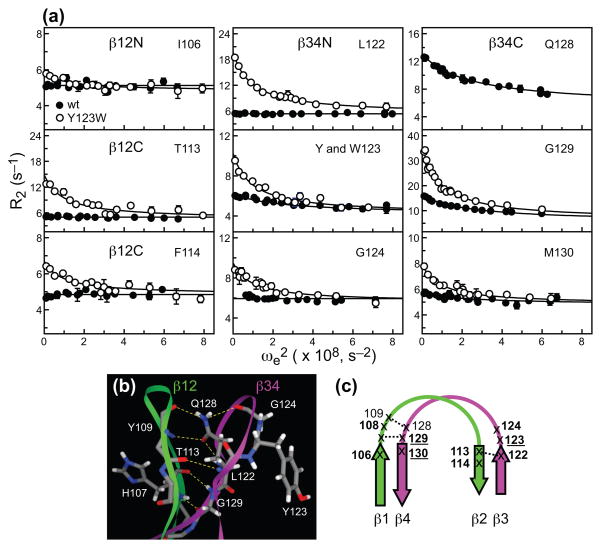
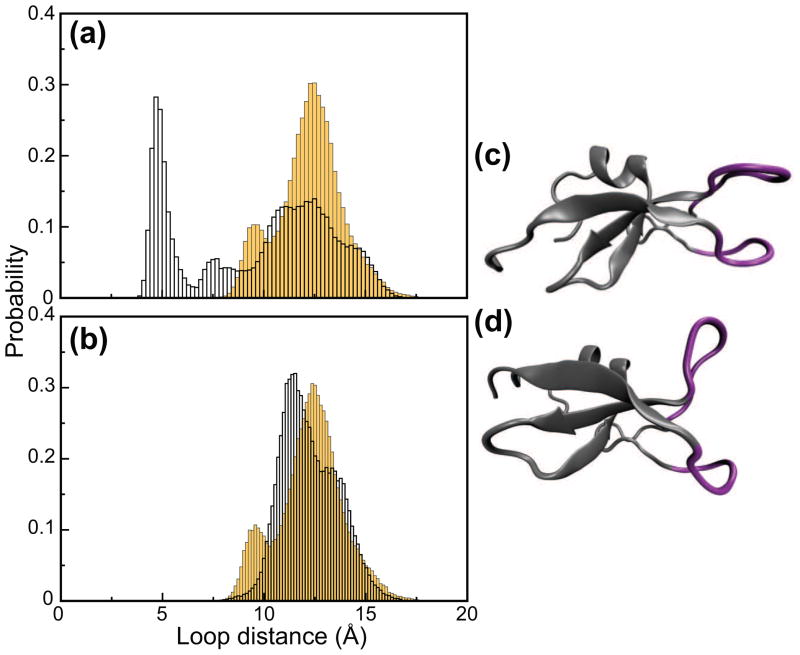
C1 domains are independently folded modules that are responsible for targeting their parent proteins to lipid membranes containing diacylglycerol (DAG), a ubiquitous second messenger. The DAG binding affinities of C1 domains determine the threshold concentration of DAG required for the propagation of signaling response and the selectivity of this response among DAG receptors in the cell. The structural information currently available for C1 domains offers little insight into the molecular basis of their differential DAG binding affinities. In this work, we characterized the C1B domain of protein kinase Cα (C1Bα) and its diagnostic mutant, Y123W, using solution NMR methods and molecular dynamics simulations. The mutation did not perturb the C1Bα structure or the sub-nanosecond dynamics of the protein backbone, but resulted in a >100-fold increase in DAG binding affinity and a substantial change in microsecond timescale conformational dynamics, as quantified by NMR rotating-frame relaxation-dispersion methods. The differences in the conformational exchange behavior between wild type and Y123W C1Bα were localized to the hinge regions of ligand-binding loops. Molecular dynamics simulations provided insight into the identity of the exchanging conformers and revealed the significance of a particular residue (Gln128) in modulating the geometry of the ligand-binding site. Taken together with the results of binding studies, our findings suggest that the conformational dynamics and preferential partitioning of the tryptophan side chain into the water-lipid interface are important factors that modulate the DAG binding properties of the C1 domains.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Interfacial partitioning of a loop hinge residue contributes to diacylglycerol affinity of conserved region 1 domains.J Biol Chem. 2014 Oct 3;289(40):27653-64. doi: 10.1074/jbc.M114.585570. Epub 2014 Aug 14. J Biol Chem. 2014. PMID: 25124034 Free PMC article.
-
Intramolecular occlusion of the diacylglycerol-binding site in the C1 domain of munc13-1.Biochemistry. 2005 Feb 1;44(4):1089-96. doi: 10.1021/bi0476127. Biochemistry. 2005. PMID: 15667202
-
Selective binding of phorbol esters and diacylglycerol by individual C1 domains of the PKD family.Biochem J. 2008 Apr 15;411(2):333-42. doi: 10.1042/BJ20071334. Biochem J. 2008. PMID: 18076381
-
C1 domains exposed: from diacylglycerol binding to protein-protein interactions.Biochim Biophys Acta. 2006 Aug;1761(8):827-37. doi: 10.1016/j.bbalip.2006.05.001. Epub 2006 May 13. Biochim Biophys Acta. 2006. PMID: 16861033 Review.
-
Regulation of the transbilayer movement of diacylglycerol in the plasma membrane.Biochimie. 2014 Dec;107 Pt A:43-50. doi: 10.1016/j.biochi.2014.09.014. Epub 2014 Sep 18. Biochimie. 2014. PMID: 25241257 Review.
Cited by
-
Reactive cysteine in the structural Zn(2+) site of the C1B domain from PKCα.Biochemistry. 2012 Sep 18;51(37):7263-77. doi: 10.1021/bi300750w. Epub 2012 Sep 5. Biochemistry. 2012. PMID: 22913772 Free PMC article.
-
Probing the Diacylglycerol Binding Site of Presynaptic Munc13-1.Biochemistry. 2021 Apr 27;60(16):1286-1298. doi: 10.1021/acs.biochem.1c00165. Epub 2021 Apr 5. Biochemistry. 2021. PMID: 33818064 Free PMC article.
-
Structural determinants of phorbol ester binding activity of the C1a and C1b domains of protein kinase C theta.Biochim Biophys Acta Biomembr. 2018 May;1860(5):1046-1056. doi: 10.1016/j.bbamem.2018.01.007. Epub 2018 Jan 6. Biochim Biophys Acta Biomembr. 2018. PMID: 29317197 Free PMC article.
-
Reactivity of Thiol-Rich Zn Sites in Diacylglycerol-Sensing PKC C1 Domain Probed by NMR Spectroscopy.Front Mol Biosci. 2021 Aug 10;8:728711. doi: 10.3389/fmolb.2021.728711. eCollection 2021. Front Mol Biosci. 2021. PMID: 34447788 Free PMC article.
-
Toggling of Diacylglycerol Affinity Correlates with Conformational Plasticity in C1 Domains.Biochemistry. 2017 May 30;56(21):2637-2640. doi: 10.1021/acs.biochem.7b00228. Epub 2017 May 18. Biochemistry. 2017. PMID: 28505428 Free PMC article.
References
-
- Brose N, Rosenmund C. Move over protein kinase C, you’ve got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. - PubMed
-
- Kazanietz MG. Targeting protein kinase C and “non-kinase” phorbol ester receptors: emerging concepts and therapeutic implications. Biochim Biophys Acta. 2005;1754:296–304. - PubMed
-
- Goel G, Makkar HP, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int J Toxicol. 2007;26:279–288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
