

AKT1/BRCA1 in the control of homologous recombination and genetic stability: the missing link between hereditary and sporadic breast cancers
- PMID: 21321378
- PMCID: PMC3157734
- DOI: 10.18632/oncotarget.203
AKT1/BRCA1 in the control of homologous recombination and genetic stability: the missing link between hereditary and sporadic breast cancers
Abstract
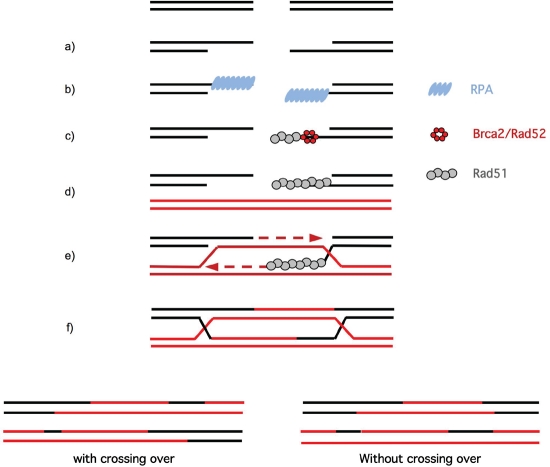
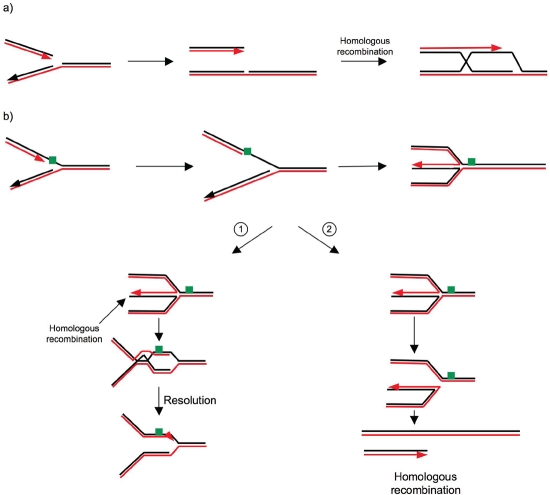
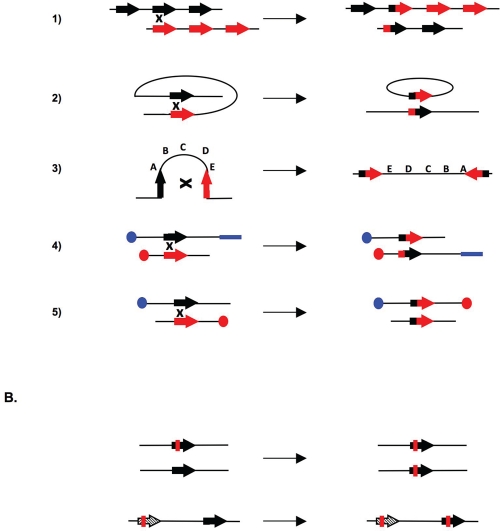
Endogenous replicative stress could be one trigger leading to tumor initiation: indeed, activation of the DNA damage response (DDR), considered the result of replicative stress, is observed in pre-cancerous cells; moreover, in hereditary breast cancers, almost all of the genes affected relate to the DDR. The most frequently mutated gene in hereditary breast cancers, BRCA1, is essential for homologous recombination (HR), a fundamental process for maintaining genome stability that permits the reactivation of blocked replication forks . Recent studies have established links between DDR and the oncogenic kinase AKT1, which is upregulated in about 50% of sporadic breast cancers. More specifically, the activation of AKT1 shows a deficient phenotype in BRCA1 and HR, revealing molecular similarities between hereditary and sporadic breast cancers. However, these results reveal a paradox regarding the physiological role of AKT1: in non-tumor cells, AKT1 promotes cellular proliferation, but consequently endangers genome integrity during replication if HR is inhibited. Since HR could itself lead to genetic instability, we propose that, under physiological conditions, moderate activation of AKT1 does not inhibit but prevents an excess of HR. The regulation of AKT1 would represent a fine transitory system for controlling HR and maintaining genomic integrity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51.Cancer Res. 2008 Nov 15;68(22):9404-12. doi: 10.1158/0008-5472.CAN-08-0861. Cancer Res. 2008. PMID: 19010915
-
Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway.Oncogene. 2013 Apr 11;32(15):1943-9. doi: 10.1038/onc.2012.211. Epub 2012 Jun 4. Oncogene. 2013. PMID: 22665067 Free PMC article.
-
Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation.Am J Hum Genet. 2006 Jun;78(6):961-72. doi: 10.1086/504090. Epub 2006 Apr 12. Am J Hum Genet. 2006. PMID: 16685647 Free PMC article.
-
Molecular functions of BRCA1 in the DNA damage response.Cancer Biol Ther. 2004 Jun;3(6):521-7. doi: 10.4161/cbt.3.6.842. Epub 2004 Jun 24. Cancer Biol Ther. 2004. PMID: 15280660 Review.
-
Therapeutic exploitation of tumor cell defects in homologous recombination.Anticancer Agents Med Chem. 2008 May;8(4):448-60. doi: 10.2174/187152008784220267. Anticancer Agents Med Chem. 2008. PMID: 18473729 Review.
Cited by
-
MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs.Cells. 2022 Apr 30;11(9):1509. doi: 10.3390/cells11091509. Cells. 2022. PMID: 35563814 Free PMC article.
-
Alteration/deficiency in activation-3 (Ada3) plays a critical role in maintaining genomic stability.Cell Cycle. 2012 Nov 15;11(22):4266-74. doi: 10.4161/cc.22613. Epub 2012 Oct 24. Cell Cycle. 2012. PMID: 23095635 Free PMC article.
-
RUNX1 and its understudied role in breast cancer.Cell Cycle. 2011 Oct 15;10(20):3461-5. doi: 10.4161/cc.10.20.18029. Epub 2011 Oct 15. Cell Cycle. 2011. PMID: 22024923 Free PMC article.
-
Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment.Cell Cycle. 2012 Oct 15;11(20):3837-50. doi: 10.4161/cc.22026. Epub 2012 Sep 14. Cell Cycle. 2012. PMID: 22983061 Free PMC article.
-
Mapping the protein-protein and genetic interactions of cancer to guide precision medicine.Curr Opin Genet Dev. 2019 Feb;54:110-117. doi: 10.1016/j.gde.2019.04.005. Epub 2019 Jul 6. Curr Opin Genet Dev. 2019. PMID: 31288129 Free PMC article. Review.
References
-
- Hyrien O. Mechanisms and consequences of replication fork arrest. Biochimie. 2000;82:5–17. - PubMed
-
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. - PubMed
-
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
