

Barth syndrome mutations that cause tafazzin complex lability
- PMID: 21300850
- PMCID: PMC3101092
- DOI: 10.1083/jcb.201008177
Barth syndrome mutations that cause tafazzin complex lability
Abstract
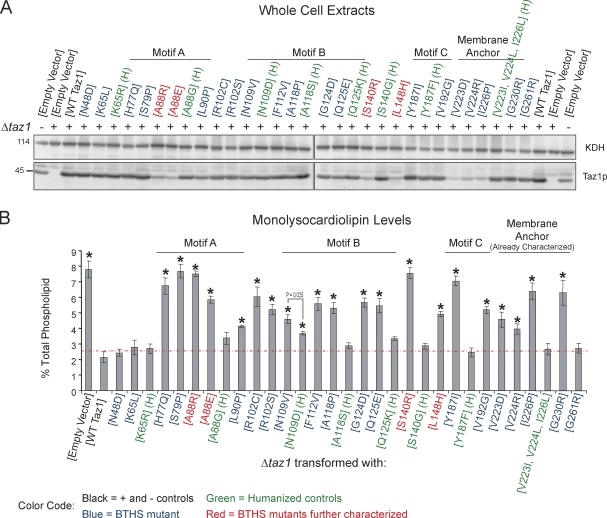
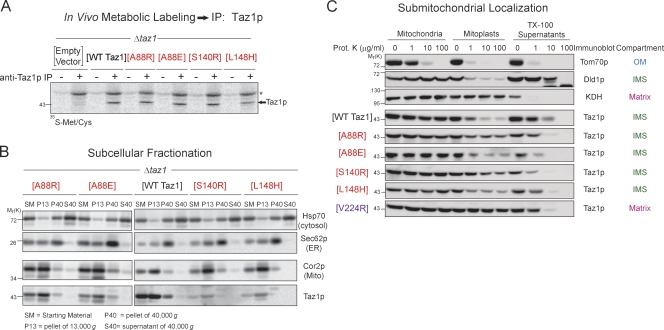
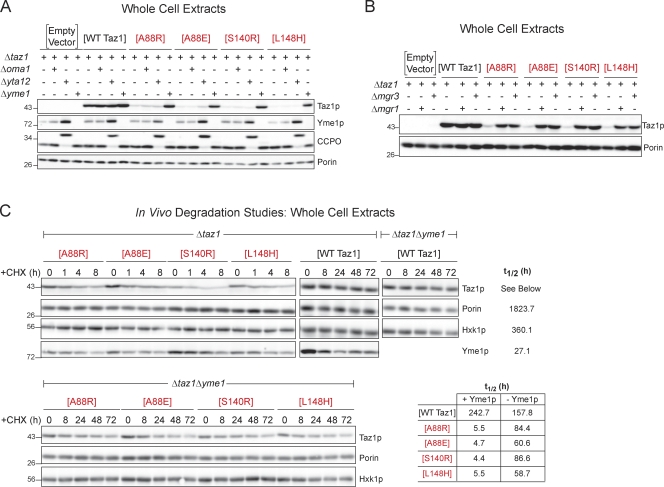
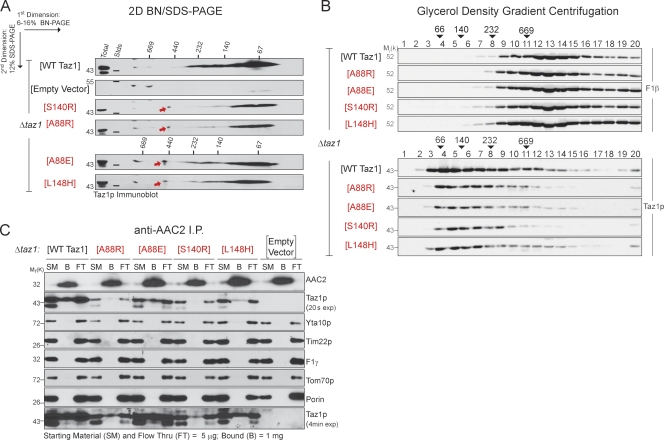
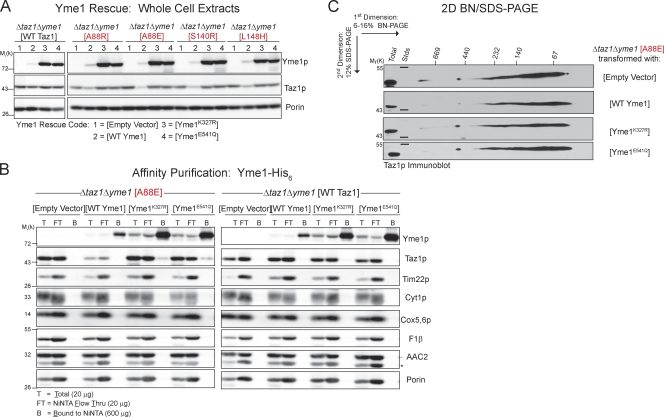
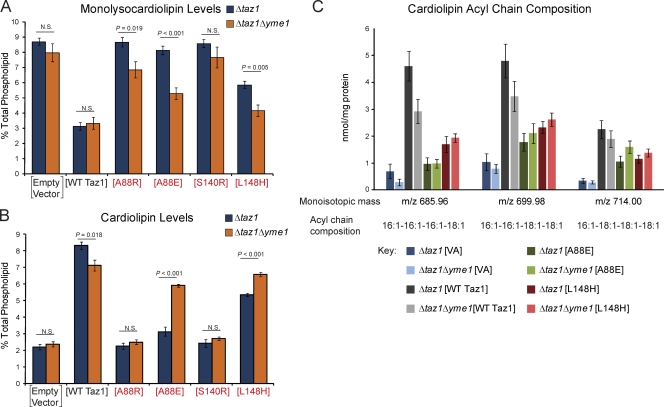
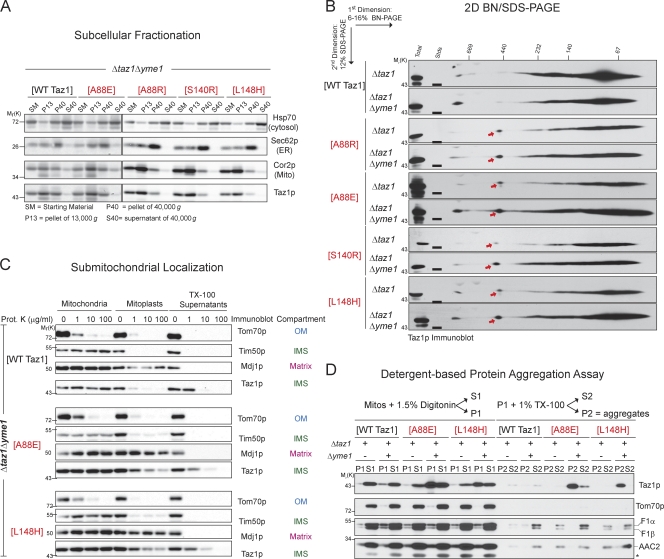
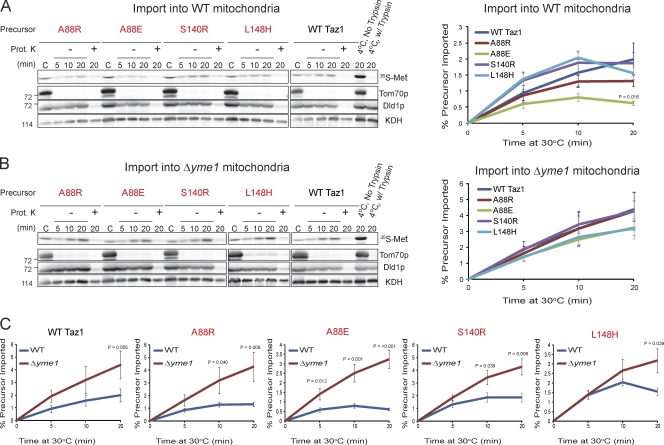
Deficits in mitochondrial function result in many human diseases. The X-linked disease Barth syndrome (BTHS) is caused by mutations in the tafazzin gene TAZ1. Its product, Taz1p, participates in the metabolism of cardiolipin, the signature phospholipid of mitochondria. In this paper, a yeast BTHS mutant tafazzin panel is established, and 18 of the 21 tested BTHS missense mutations cannot functionally replace endogenous tafazzin. Four BTHS mutant tafazzins expressed at low levels are degraded by the intermembrane space AAA (i-AAA) protease, suggesting misfolding of the mutant polypeptides. Paradoxically, each of these mutant tafazzins assembles in normal protein complexes. Furthermore, in the absence of the i-AAA protease, increased expression and assembly of two of the BTHS mutants improve their function. However, the BTHS mutant complexes are extremely unstable and accumulate as insoluble aggregates when disassembled in the absence of the i-AAA protease. Thus, the loss of function for these BTHS mutants results from the inherent instability of the mutant tafazzin complexes.
Figures

Similar articles
-
Seven functional classes of Barth syndrome mutation.Hum Mol Genet. 2013 Feb 1;22(3):483-92. doi: 10.1093/hmg/dds447. Epub 2012 Oct 24. Hum Mol Genet. 2013. PMID: 23100323 Free PMC article.
-
Overexpression of branched-chain amino acid aminotransferases rescues the growth defects of cells lacking the Barth syndrome-related gene TAZ1.J Mol Med (Berl). 2019 Feb;97(2):269-279. doi: 10.1007/s00109-018-1728-4. Epub 2019 Jan 3. J Mol Med (Berl). 2019. PMID: 30604168
-
Cardiolipin function in the yeast S. cerevisiae and the lessons learned for Barth syndrome.J Inherit Metab Dis. 2022 Jan;45(1):60-71. doi: 10.1002/jimd.12447. Epub 2021 Oct 19. J Inherit Metab Dis. 2022. PMID: 34626131 Free PMC article. Review.
-
Defining functional classes of Barth syndrome mutation in humans.Hum Mol Genet. 2016 May 1;25(9):1754-70. doi: 10.1093/hmg/ddw046. Epub 2016 Feb 16. Hum Mol Genet. 2016. PMID: 26908608 Free PMC article.
-
Barth Syndrome Cardiomyopathy: An Update.Genes (Basel). 2022 Apr 8;13(4):656. doi: 10.3390/genes13040656. Genes (Basel). 2022. PMID: 35456462 Free PMC article. Review.
Cited by
-
Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies.Front Genet. 2016 Jan 20;6:359. doi: 10.3389/fgene.2015.00359. eCollection 2015. Front Genet. 2016. PMID: 26834781 Free PMC article. Review.
-
Studying Lipid-Related Pathophysiology Using the Yeast Model.Front Physiol. 2021 Oct 28;12:768411. doi: 10.3389/fphys.2021.768411. eCollection 2021. Front Physiol. 2021. PMID: 34777024 Free PMC article. Review.
-
Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach.J Multidiscip Healthc. 2015 Jul 29;8:345-58. doi: 10.2147/JMDH.S54802. eCollection 2015. J Multidiscip Healthc. 2015. PMID: 26251611 Free PMC article. Review.
-
Proteolytic Control of Lipid Metabolism.ACS Chem Biol. 2019 Nov 15;14(11):2406-2423. doi: 10.1021/acschembio.9b00695. Epub 2019 Sep 30. ACS Chem Biol. 2019. PMID: 31503446 Free PMC article. Review.
-
Disentangling oxidation/hydrolysis reactions of brain mitochondrial cardiolipins in pathogenesis of traumatic injury.JCI Insight. 2018 Nov 2;3(21):e97677. doi: 10.1172/jci.insight.97677. JCI Insight. 2018. PMID: 30385716 Free PMC article.
References
-
- Barth P.G., Scholte H.R., Berden J.A., Van der Klei-Van Moorsel J.M., Luyt-Houwen I.E., Van’t Veer-Korthof E.T., Van der Harten J.J., Sobotka-Plojhar M.A. 1983. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62:327–355 10.1016/0022-510X(83)90209-5 - DOI - PubMed
-
- Barth P.G., Van den Bogert C., Bolhuis P.A., Scholte H.R., van Gennip A.H., Schutgens R.B., Ketel A.G. 1996. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 19:157–160 10.1007/BF01799418 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
