

Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy
- PMID: 21296869
- PMCID: PMC3071670
- DOI: 10.1093/hmg/ddr048
Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy
Abstract
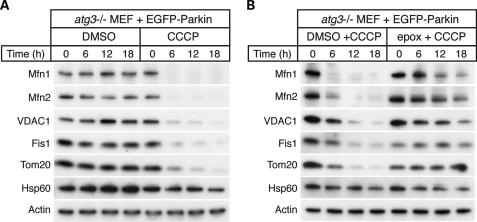
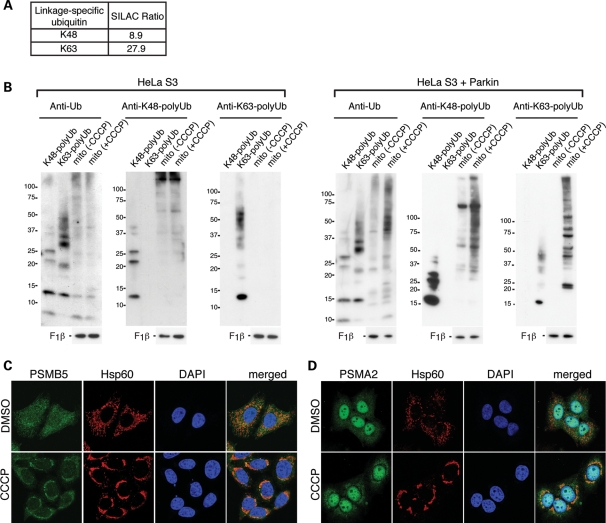
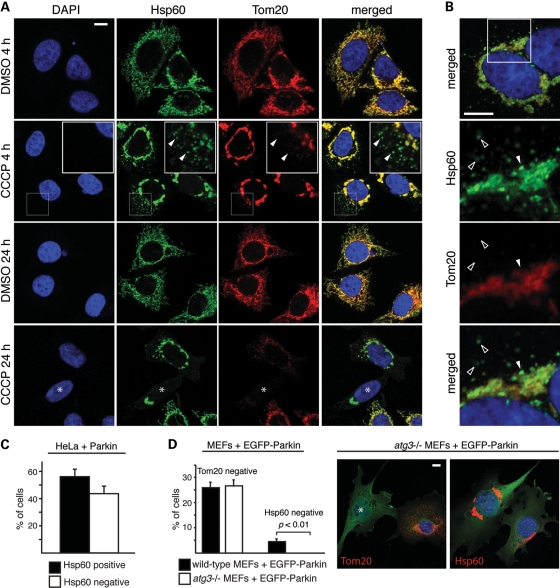
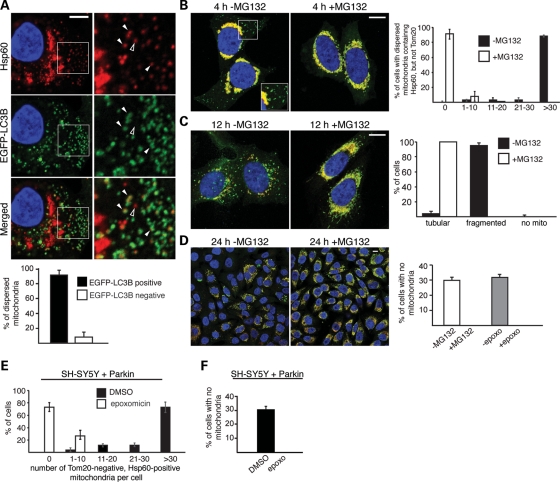
Parkin, an E3 ubiquitin ligase implicated in Parkinson's disease, promotes degradation of dysfunctional mitochondria by autophagy. Using proteomic and cellular approaches, we show that upon translocation to mitochondria, Parkin activates the ubiquitin-proteasome system (UPS) for widespread degradation of outer membrane proteins. This is evidenced by an increase in K48-linked polyubiquitin on mitochondria, recruitment of the 26S proteasome and rapid degradation of multiple outer membrane proteins. The degradation of proteins by the UPS occurs independently of the autophagy pathway, and inhibition of the 26S proteasome completely abrogates Parkin-mediated mitophagy in HeLa, SH-SY5Y and mouse cells. Although the mitofusins Mfn1 and Mfn2 are rapid degradation targets of Parkin, we find that degradation of additional targets is essential for mitophagy. These results indicate that remodeling of the mitochondrial outer membrane proteome is important for mitophagy, and reveal a causal link between the UPS and autophagy, the major pathways for degradation of intracellular substrates.
Figures

Similar articles
-
Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin.J Cell Biol. 2010 Dec 27;191(7):1367-80. doi: 10.1083/jcb.201007013. Epub 2010 Dec 20. J Cell Biol. 2010. PMID: 21173115 Free PMC article.
-
Parkin uses the UPS to ship off dysfunctional mitochondria.Autophagy. 2011 Jul;7(7):771-2. doi: 10.4161/auto.7.7.15453. Epub 2011 Jul 1. Autophagy. 2011. PMID: 21460629 Free PMC article.
-
N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase.Curr Genet. 2020 Aug;66(4):693-701. doi: 10.1007/s00294-020-01062-2. Epub 2020 Mar 10. Curr Genet. 2020. PMID: 32157382 Review.
-
Inhibition of proteasome reveals basal mitochondrial ubiquitination.J Proteomics. 2020 Oct 30;229:103949. doi: 10.1016/j.jprot.2020.103949. Epub 2020 Aug 31. J Proteomics. 2020. PMID: 32882436
-
Beyond Deubiquitylation: USP30-Mediated Regulation of Mitochondrial Homeostasis.Adv Exp Med Biol. 2017;1038:133-148. doi: 10.1007/978-981-10-6674-0_10. Adv Exp Med Biol. 2017. PMID: 29178074 Review.
Cited by
-
The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo.Proc Natl Acad Sci U S A. 2013 Apr 16;110(16):6400-5. doi: 10.1073/pnas.1221132110. Epub 2013 Mar 18. Proc Natl Acad Sci U S A. 2013. PMID: 23509287 Free PMC article.
-
Integrating pathways of Parkinson's disease in a molecular interaction map.Mol Neurobiol. 2014 Feb;49(1):88-102. doi: 10.1007/s12035-013-8489-4. Epub 2013 Jul 7. Mol Neurobiol. 2014. PMID: 23832570 Free PMC article. Review.
-
Cardiac aging: from molecular mechanisms to significance in human health and disease.Antioxid Redox Signal. 2012 Jun 15;16(12):1492-526. doi: 10.1089/ars.2011.4179. Epub 2012 Apr 3. Antioxid Redox Signal. 2012. PMID: 22229339 Free PMC article. Review.
-
Electron microscopic analysis of a spherical mitochondrial structure.J Biol Chem. 2012 Dec 7;287(50):42373-8. doi: 10.1074/jbc.M112.413674. Epub 2012 Oct 23. J Biol Chem. 2012. PMID: 23093403 Free PMC article.
-
Mitochondrial protein dysfunction in pathogenesis of neurological diseases.Front Mol Neurosci. 2022 Sep 7;15:974480. doi: 10.3389/fnmol.2022.974480. eCollection 2022. Front Mol Neurosci. 2022. PMID: 36157077 Free PMC article. Review.
References
-
- Clark I.E., Dodson M.W., Jiang C., Cao J.H., Huh J.R., Seol J.H., Yoo S.J., Hay B.A., Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi:10.1038/nature04779. - DOI - PubMed
-
- Greene J.C., Whitworth A.J., Kuo I., Andrews L.A., Feany M.B., Pallanck L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA. 2003;100:4078–4083. doi:10.1073/pnas.0737556100. - DOI - PMC - PubMed
-
- Palacino J.J., Sagi D., Goldberg M.S., Krauss S., Motz C., Wacker M., Klose J., Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004;279:18614–18622. doi:10.1074/jbc.M401135200. - DOI - PubMed
-
- Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.M., et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi:10.1038/nature04788. - DOI - PubMed
-
- Gautier C.A., Kitada T., Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl Acad. Sci. USA. 2008;105:11364–11369. doi:10.1073/pnas.0802076105. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
