

Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture
- PMID: 21292630
- PMCID: PMC3068000
- DOI: 10.1093/gbe/evr007
Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture
Abstract
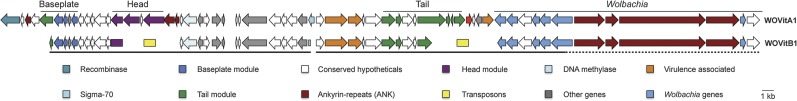
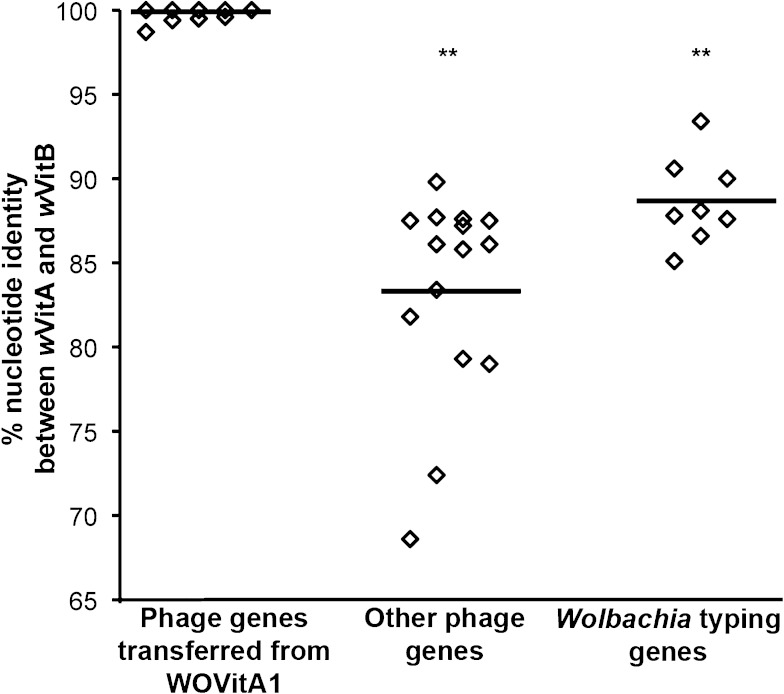
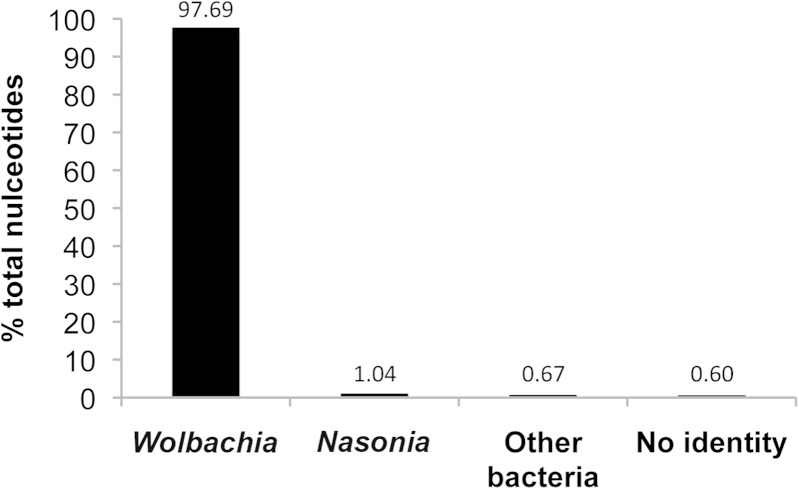
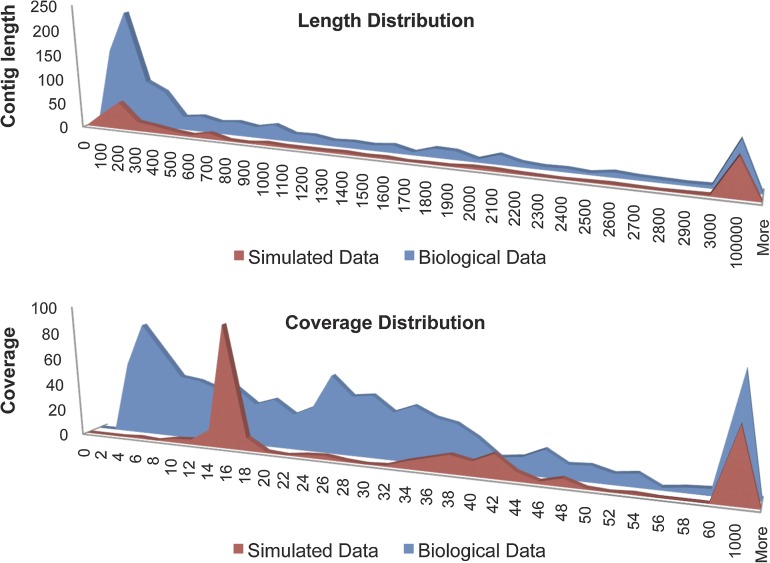
Bacteriophage flux can cause the majority of genetic diversity in free-living bacteria. This tenet of bacterial genome evolution generally does not extend to obligate intracellular bacteria owing to their reduced contact with other microbes and a predominance of gene deletion over gene transfer. However, recent studies suggest intracellular coinfections in the same host can facilitate exchange of mobile elements between obligate intracellular bacteria-a means by which these bacteria can partially mitigate the reductive forces of the intracellular lifestyle. To test whether bacteriophages transfer as single genes or larger regions between coinfections, we sequenced the genome of the obligate intracellular Wolbachia strain wVitB from the parasitic wasp Nasonia vitripennis and compared it against the prophage sequences of the divergent wVitA coinfection. We applied, for the first time, a targeted sequence capture array to specifically trap the symbiont's DNA from a heterogeneous mixture of eukaryotic, bacterial, and viral DNA. The tiled array successfully captured the genome with 98.3% efficiency. Examination of the genome sequence revealed the largest transfer of bacteriophage and flanking genes (52.2 kb) to date between two obligate intracellular coinfections. The mobile element transfer occurred in the recent evolutionary past based on the 99.9% average nucleotide identity of the phage sequences between the two strains. In addition to discovering an evolutionary recent and large-scale horizontal phage transfer between coinfecting obligate intracellular bacteria, we demonstrate that "targeted genome capture" can enrich target DNA to alleviate the problem of isolating symbiotic microbes that are difficult to culture or purify from the conglomerate of organisms inside eukaryotes.
Figures
Similar articles
-
Evolutionary genomics of a temperate bacteriophage in an obligate intracellular bacteria (Wolbachia).PLoS One. 2011;6(9):e24984. doi: 10.1371/journal.pone.0024984. Epub 2011 Sep 14. PLoS One. 2011. PMID: 21949820 Free PMC article.
-
Phage WO of Wolbachia: lambda of the endosymbiont world.Trends Microbiol. 2010 Apr;18(4):173-81. doi: 10.1016/j.tim.2009.12.011. Epub 2010 Jan 18. Trends Microbiol. 2010. PMID: 20083406 Free PMC article. Review.
-
Lateral phage transfer in obligate intracellular bacteria (wolbachia): verification from natural populations.Mol Biol Evol. 2010 Mar;27(3):501-5. doi: 10.1093/molbev/msp275. Epub 2009 Nov 11. Mol Biol Evol. 2010. PMID: 19906794 Free PMC article.
-
Isolation and characterization of the bacteriophage WO from Wolbachia, an arthropod endosymbiont.Biochem Biophys Res Commun. 2004 May 14;317(4):1183-8. doi: 10.1016/j.bbrc.2004.03.164. Biochem Biophys Res Commun. 2004. PMID: 15094394
-
Wolbachia genomes: insights into an intracellular lifestyle.Curr Biol. 2005 Jul 12;15(13):R507-9. doi: 10.1016/j.cub.2005.06.029. Curr Biol. 2005. PMID: 16005284 Review.
Cited by
-
What viruses tell us about evolution and immunity: beyond Darwin?Ann N Y Acad Sci. 2019 Jul;1447(1):53-68. doi: 10.1111/nyas.14097. Epub 2019 Apr 29. Ann N Y Acad Sci. 2019. PMID: 31032941 Free PMC article. Review.
-
Widespread phages of endosymbionts: Phage WO genomics and the proposed taxonomic classification of Symbioviridae.PLoS Genet. 2022 Jun 6;18(6):e1010227. doi: 10.1371/journal.pgen.1010227. eCollection 2022 Jun. PLoS Genet. 2022. PMID: 35666732 Free PMC article.
-
Bacteriophage WO Can Mediate Horizontal Gene Transfer in Endosymbiotic Wolbachia Genomes.Front Microbiol. 2016 Nov 29;7:1867. doi: 10.3389/fmicb.2016.01867. eCollection 2016. Front Microbiol. 2016. PMID: 27965627 Free PMC article.
-
A systems biology approach for studying Wolbachia metabolism reveals points of interaction with its host in the context of arboviral infection.PLoS Negl Trop Dis. 2019 Aug 30;13(8):e0007678. doi: 10.1371/journal.pntd.0007678. eCollection 2019 Aug. PLoS Negl Trop Dis. 2019. PMID: 31469838 Free PMC article.
-
Complete de novo assembly of Wolbachia endosymbiont of Diaphorina citri Kuwayama (Hemiptera: Liviidae) using long-read genome sequencing.Sci Rep. 2022 Jan 7;12(1):125. doi: 10.1038/s41598-021-03184-0. Sci Rep. 2022. PMID: 34996906 Free PMC article.
References
-
- Akman L, et al. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet. 2002;32:402–407. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Andersson SG, et al. Comparative genomics of microbial pathogens and symbionts. Bioinformatics. 2002;18(2 Suppl):S17. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
