

Bim and Mcl-1 exert key roles in regulating JAK2V617F cell survival
- PMID: 21247487
- PMCID: PMC3037340
- DOI: 10.1186/1471-2407-11-24
Bim and Mcl-1 exert key roles in regulating JAK2V617F cell survival
Abstract
Background: The JAK2V617F mutation plays a major role in the pathogenesis of myeloproliferative neoplasms and is found in the vast majority of patients suffering from polycythemia vera and in roughly every second patient suffering from essential thrombocythemia or from primary myelofibrosis. The V617F mutation is thought to provide hematopoietic stem cells and myeloid progenitors with a survival and proliferation advantage. It has previously been shown that activated JAK2 promotes cell survival by upregulating the anti-apoptotic STAT5 target gene Bcl-xL. In this study, we have investigated the role of additional apoptotic players, the pro-apoptotic protein Bim as well as the anti-apoptotic protein Mcl-1.
Methods: Pharmacological inhibition of JAK2/STAT5 signaling in JAK2V617F mutant SET-2 and MB-02 cells was used to study effects on signaling, cell proliferation and apoptosis by Western blot analysis, WST-1 proliferation assays and flow cytometry. Cells were transfected with siRNA oligos to deplete candidate pro- and anti-apoptotic proteins. Co-immunoprecipitation assays were performed to assess the impact of JAK2 inhibition on complexes of pro- and anti-apoptotic proteins.
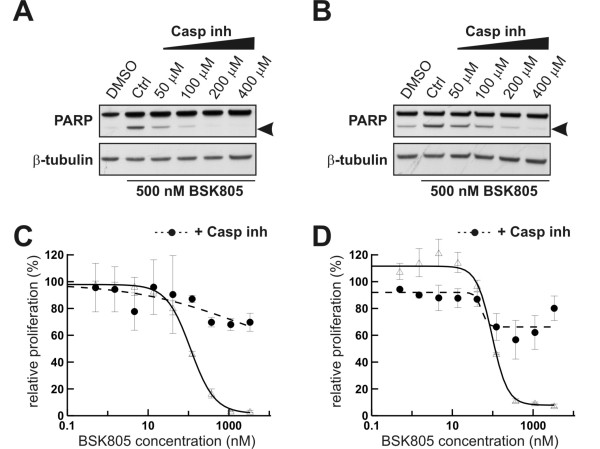
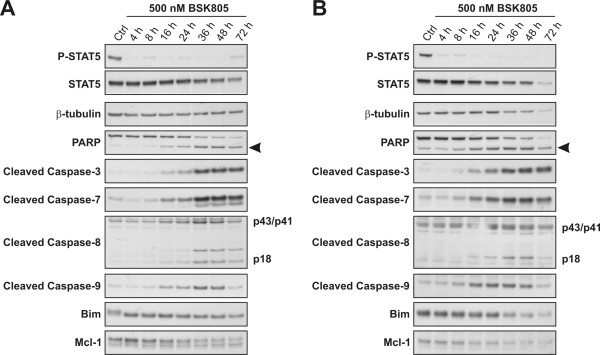
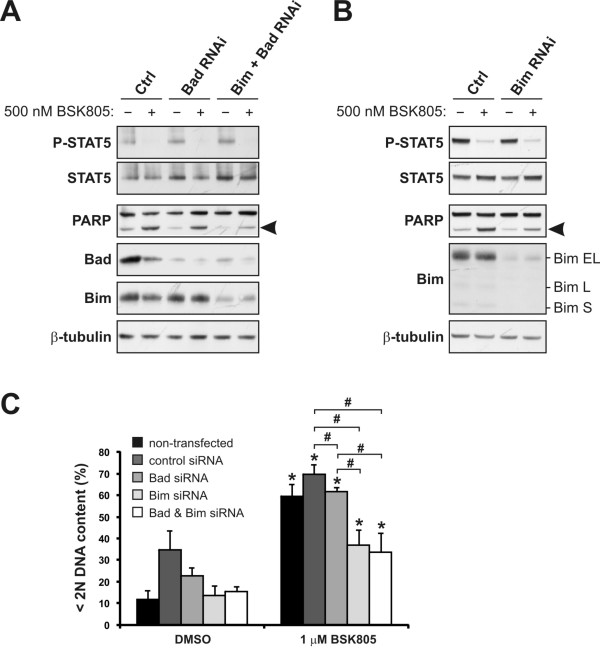
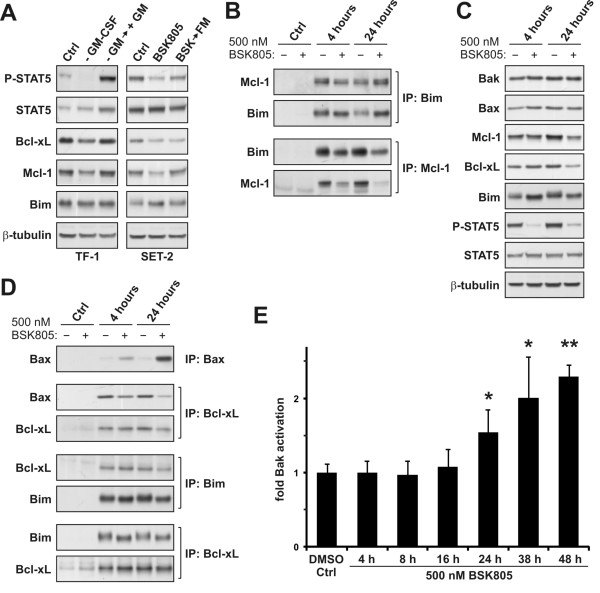
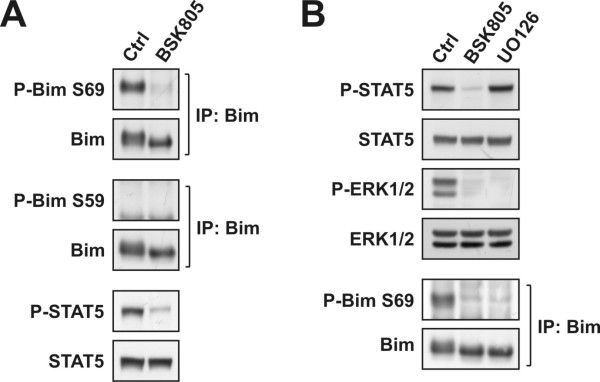
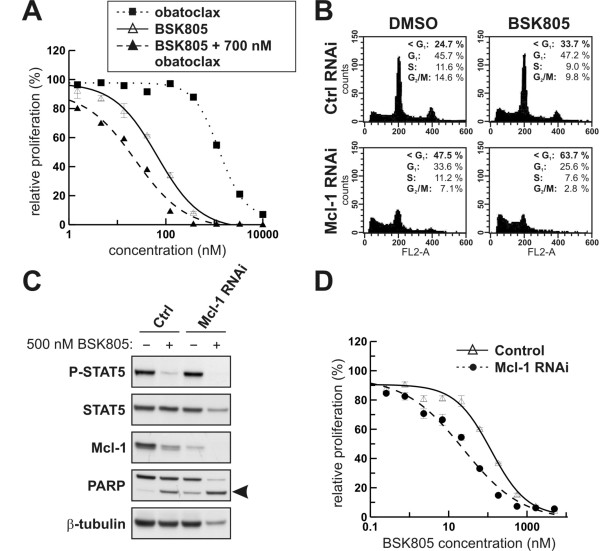
Results: Treatment of JAK2V617F mutant cell lines with a JAK2 inhibitor was found to trigger Bim activation. Furthermore, Bim depletion by RNAi suppressed JAK2 inhibitor-induced cell death. Bim activation following JAK2 inhibition led to enhanced sequestration of Mcl-1, besides Bcl-xL. Importantly, Mcl-1 depletion by RNAi was sufficient to compromise JAK2V617F mutant cell viability and sensitized the cells to JAK2 inhibition.
Conclusions: We conclude that Bim and Mcl-1 have key opposing roles in regulating JAK2V617F cell survival and propose that inactivation of aberrant JAK2 signaling leads to changes in Bim complexes that trigger cell death. Thus, further preclinical evaluation of combinations of JAK2 inhibitors with Bcl-2 family antagonists that also tackle Mcl-1, besides Bcl-xL, is warranted to assess the therapeutic potential for the treatment of chronic myeloproliferative neoplasms.
Figures

Similar articles
-
c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1.Mol Cancer. 2011 Sep 7;10:110. doi: 10.1186/1476-4598-10-110. Mol Cancer. 2011. PMID: 21899728 Free PMC article.
-
Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis.J Biol Chem. 2006 Apr 14;281(15):10153-63. doi: 10.1074/jbc.M510349200. Epub 2006 Feb 13. J Biol Chem. 2006. PMID: 16478725
-
Up-regulation of pro-apoptotic protein Bim and down-regulation of anti-apoptotic protein Mcl-1 cooperatively mediate enhanced tumor cell death induced by the combination of ERK kinase (MEK) inhibitor and microtubule inhibitor.J Biol Chem. 2012 Mar 23;287(13):10289-10300. doi: 10.1074/jbc.M111.319426. Epub 2012 Jan 23. J Biol Chem. 2012. PMID: 22270368 Free PMC article.
-
Bim and the pro-survival Bcl-2 proteins: opposites attract, ERK repels.Cell Cycle. 2007 Sep 15;6(18):2236-40. doi: 10.4161/cc.6.18.4728. Epub 2007 Jul 10. Cell Cycle. 2007. PMID: 17881896 Review.
-
Regulation of Bim in Health and Disease.Oncotarget. 2015 Sep 15;6(27):23058-134. doi: 10.18632/oncotarget.5492. Oncotarget. 2015. PMID: 26405162 Free PMC article. Review.
Cited by
-
Suppression of multiple anti-apoptotic BCL2 family proteins recapitulates the effects of JAK2 inhibitors in JAK2V617F driven myeloproliferative neoplasms.Cancer Sci. 2022 Feb;113(2):597-608. doi: 10.1111/cas.15210. Epub 2021 Dec 15. Cancer Sci. 2022. PMID: 34808021 Free PMC article.
-
Mechanisms for mTORC1 activation and synergistic induction of apoptosis by ruxolitinib and BH3 mimetics or autophagy inhibitors in JAK2-V617F-expressing leukemic cells including newly established PVTL-2.Oncotarget. 2018 Jun 1;9(42):26834-26851. doi: 10.18632/oncotarget.25515. eCollection 2018 Jun 1. Oncotarget. 2018. PMID: 29928488 Free PMC article.
-
JAK2V617F drives Mcl-1 expression and sensitizes hematologic cell lines to dual inhibition of JAK2 and Bcl-xL.PLoS One. 2015 Mar 17;10(3):e0114363. doi: 10.1371/journal.pone.0114363. eCollection 2015. PLoS One. 2015. PMID: 25781882 Free PMC article.
-
Identification of myeloproliferative neoplasm drug agents via predictive simulation modeling: assessing responsiveness with micro-environment derived cytokines.Oncotarget. 2016 Jun 14;7(24):35989-36001. doi: 10.18632/oncotarget.8540. Oncotarget. 2016. PMID: 27056884 Free PMC article.
-
Exploration and identification of anoikis-related genes in polycythemia vera.Front Genet. 2023 Feb 17;14:1139351. doi: 10.3389/fgene.2023.1139351. eCollection 2023. Front Genet. 2023. PMID: 36873934 Free PMC article.
References
-
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, Boggon TJ, Wlodarska I, Clark JJ, Moore S. et al.Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
