

Experimental models of Rett syndrome based on Mecp2 dysfunction
- PMID: 21239731
- PMCID: PMC3059199
- DOI: 10.1258/ebm.2010.010261
Experimental models of Rett syndrome based on Mecp2 dysfunction
Abstract
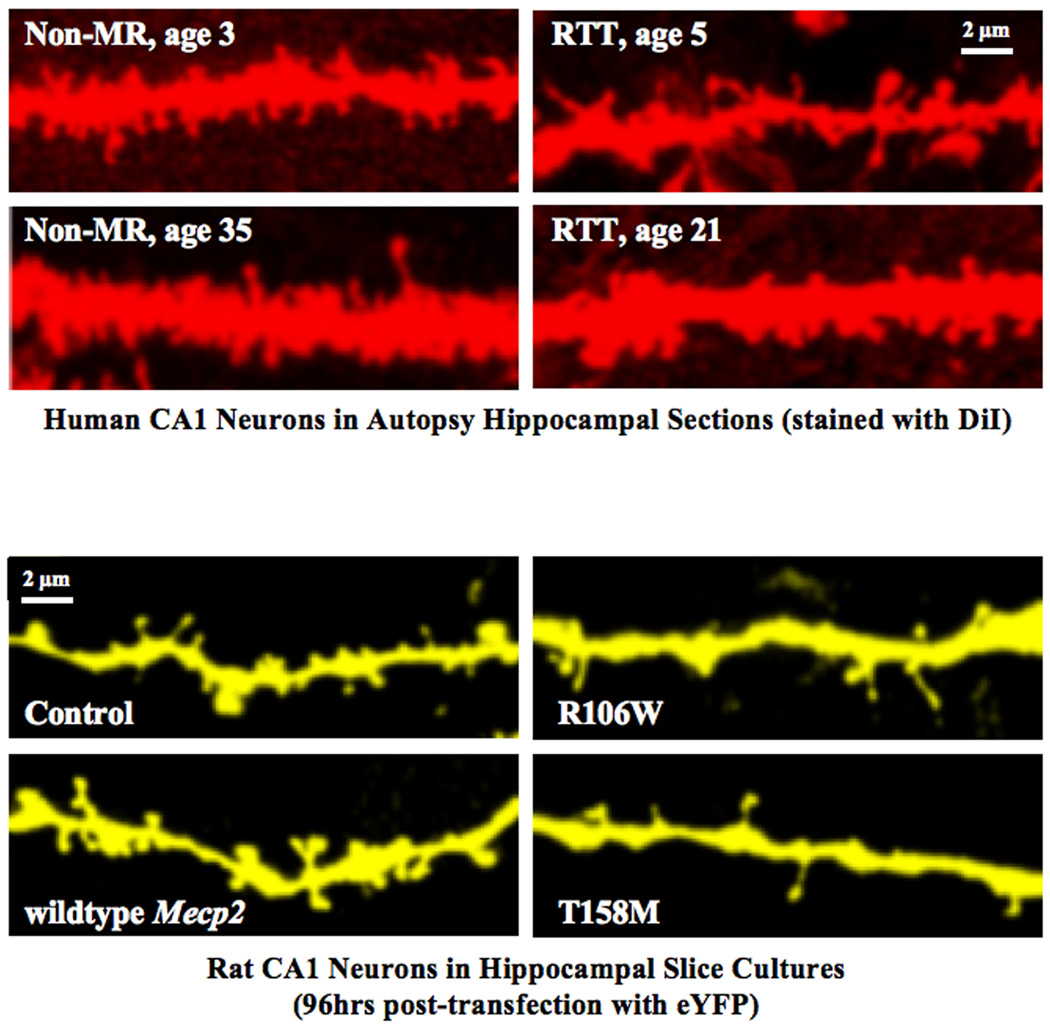
Rett syndrome (RTT) is a neurodevelopmental disorder predominantly occurring in females with an incidence of 1:10,000 births and caused by sporadic mutations in the MECP2 gene, which encodes methyl-CpG-binding protein-2, an epigenetic transcription factor that binds methylated DNA. The clinical hallmarks include a period of apparently normal early development followed by a plateau and then subsequent frank regression. Impaired visual and aural contact often lead to an initial diagnosis of autism. The characterization of experimental models based on the loss-of-function of the mouse Mecp2 gene revealed that subtle changes in the morphology and function of brain cells and synapses have profound consequences on network activities that underlie critical brain functions. Furthermore, these experimental models have been used for successful reversals of RTT-like symptoms by genetic, pharmacological and environmental manipulations, raising hope for novel therapeutic strategies to improve the quality of life of RTT individuals.
Figures
Similar articles
-
Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome.Free Radic Biol Med. 2015 Nov;88(Pt A):81-90. doi: 10.1016/j.freeradbiomed.2015.04.019. Epub 2015 May 8. Free Radic Biol Med. 2015. PMID: 25960047 Review.
-
MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome.Proc Natl Acad Sci U S A. 2015 Apr 28;112(17):5509-14. doi: 10.1073/pnas.1505909112. Epub 2015 Apr 13. Proc Natl Acad Sci U S A. 2015. PMID: 25870282 Free PMC article.
-
Adult neural function requires MeCP2.Science. 2011 Jul 8;333(6039):186. doi: 10.1126/science.1206593. Epub 2011 Jun 2. Science. 2011. PMID: 21636743 Free PMC article.
-
Rett syndrome and MeCP2.Neuromolecular Med. 2014 Jun;16(2):231-64. doi: 10.1007/s12017-014-8295-9. Epub 2014 Mar 11. Neuromolecular Med. 2014. PMID: 24615633 Free PMC article. Review.
-
Rett syndrome: a complex disorder with simple roots.Nat Rev Genet. 2015 May;16(5):261-75. doi: 10.1038/nrg3897. Epub 2015 Mar 3. Nat Rev Genet. 2015. PMID: 25732612 Review.
Cited by
-
Preclinical research in Rett syndrome: setting the foundation for translational success.Dis Model Mech. 2012 Nov;5(6):733-45. doi: 10.1242/dmm.011007. Dis Model Mech. 2012. PMID: 23115203 Free PMC article. Review.
-
Evaluation of current pharmacological treatment options in the management of Rett syndrome: from the present to future therapeutic alternatives.Curr Clin Pharmacol. 2013 Nov;8(4):358-69. doi: 10.2174/15748847113086660069. Curr Clin Pharmacol. 2013. PMID: 24050745 Free PMC article. Review.
-
JNK signaling provides a novel therapeutic target for Rett syndrome.BMC Biol. 2021 Dec 16;19(1):256. doi: 10.1186/s12915-021-01190-2. BMC Biol. 2021. PMID: 34911542 Free PMC article.
-
Characterization of Rett Syndrome-like phenotypes in Mecp2-knockout rats.J Neurodev Disord. 2016 Jun 16;8:23. doi: 10.1186/s11689-016-9156-7. eCollection 2016. J Neurodev Disord. 2016. PMID: 27313794 Free PMC article.
-
Autism Spectrum Disorder and miRNA: An Overview of Experimental Models.Brain Sci. 2019 Oct 3;9(10):265. doi: 10.3390/brainsci9100265. Brain Sci. 2019. PMID: 31623367 Free PMC article. Review.
References
-
- Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood] Wien Med Wochenschr. 1966;116(37):723–726. - PubMed
-
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471–479. - PubMed
-
- Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, Williamson S, Leonard H. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148(3):347–352. - PubMed
-
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–437. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
