

Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair
- PMID: 21209706
- PMCID: PMC3010660
- DOI: 10.4061/2010/592980
Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair
Abstract
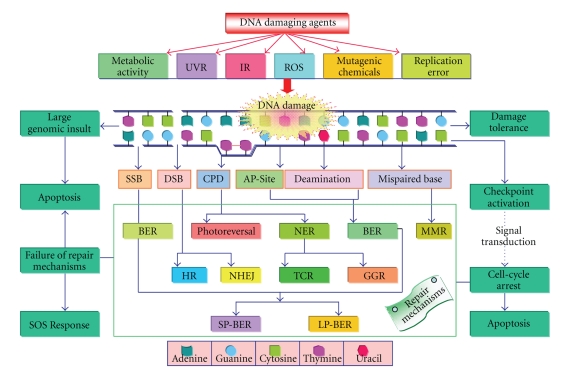
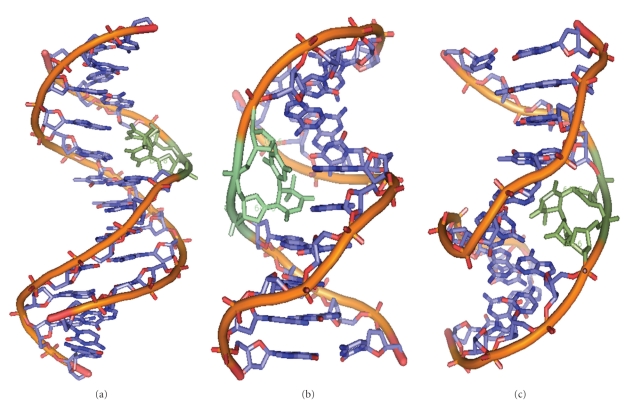
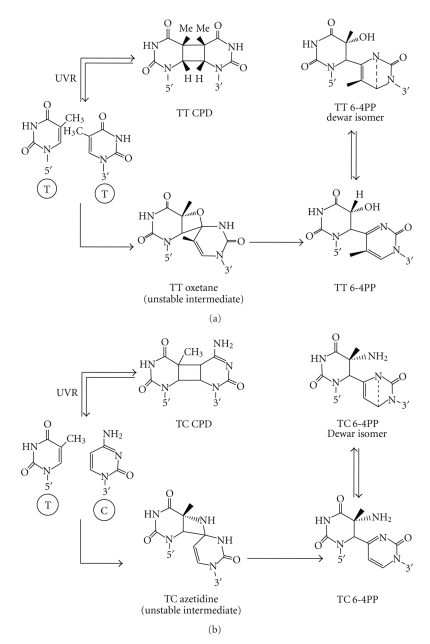
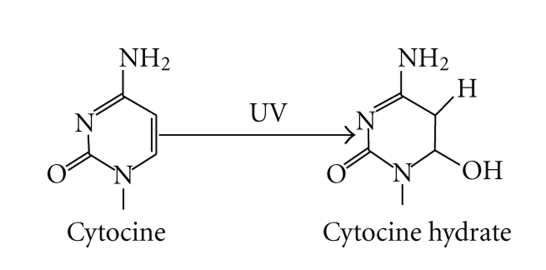
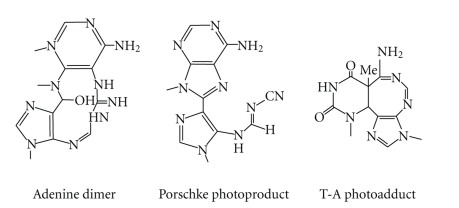
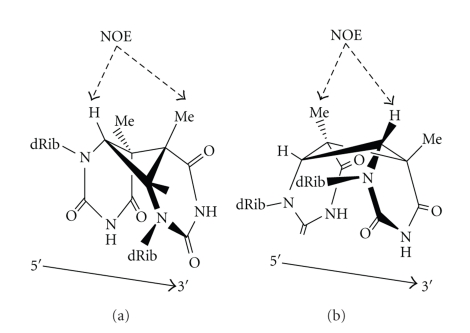
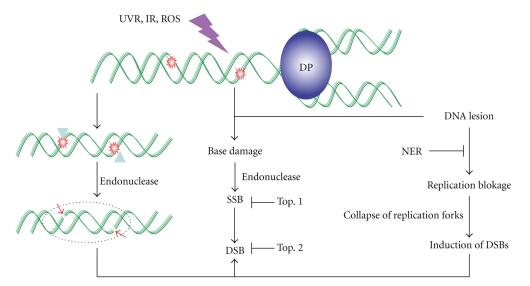
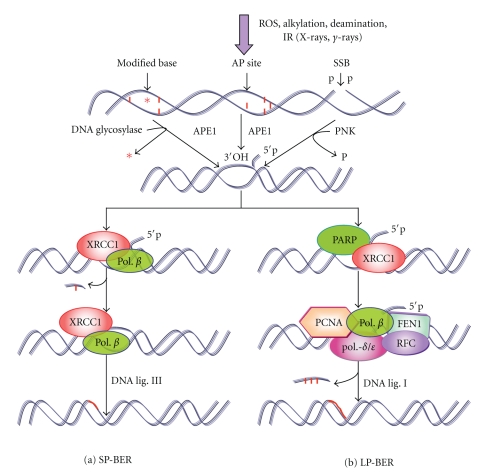
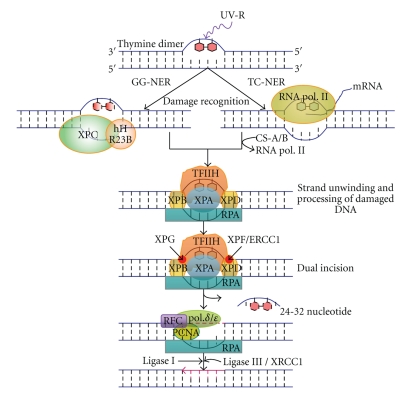
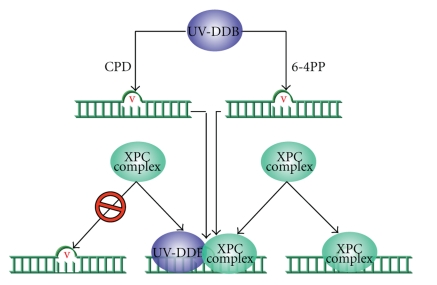
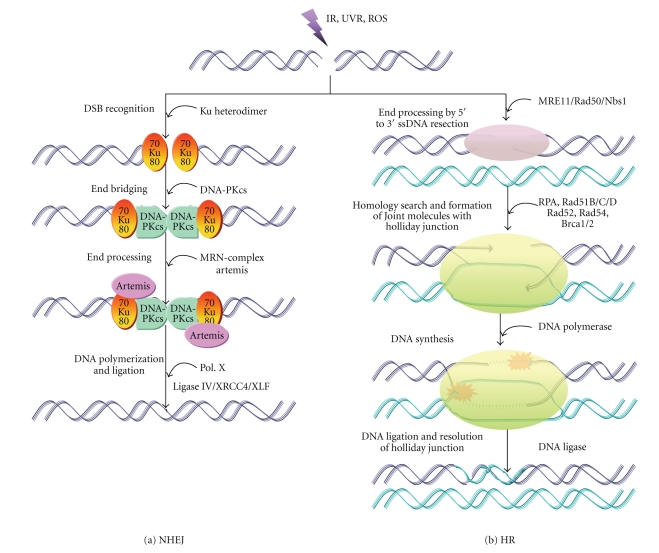
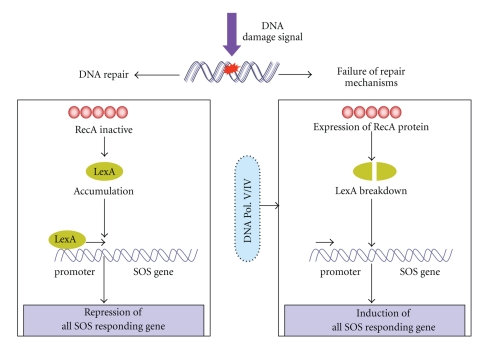
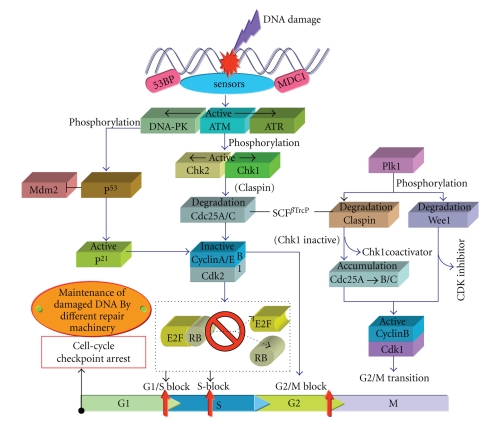
DNA is one of the prime molecules, and its stability is of utmost importance for proper functioning and existence of all living systems. Genotoxic chemicals and radiations exert adverse effects on genome stability. Ultraviolet radiation (UVR) (mainly UV-B: 280-315 nm) is one of the powerful agents that can alter the normal state of life by inducing a variety of mutagenic and cytotoxic DNA lesions such as cyclobutane-pyrimidine dimers (CPDs), 6-4 photoproducts (6-4PPs), and their Dewar valence isomers as well as DNA strand breaks by interfering the genome integrity. To counteract these lesions, organisms have developed a number of highly conserved repair mechanisms such as photoreactivation, base excision repair (BER), nucleotide excision repair (NER), and mismatch repair (MMR). Additionally, double-strand break repair (by homologous recombination and nonhomologous end joining), SOS response, cell-cycle checkpoints, and programmed cell death (apoptosis) are also operative in various organisms with the expense of specific gene products. This review deals with UV-induced alterations in DNA and its maintenance by various repair mechanisms.
Figures


Similar articles
-
Physiological aspects of UV-excitation of DNA.Top Curr Chem. 2015;356:203-48. doi: 10.1007/128_2014_531. Top Curr Chem. 2015. PMID: 24696352 Review.
-
UV-induced DNA damage and repair: a review.Photochem Photobiol Sci. 2002 Apr;1(4):225-36. doi: 10.1039/b201230h. Photochem Photobiol Sci. 2002. PMID: 12661961 Review.
-
Solar ultraviolet radiation-induced DNA damage in aquatic organisms: potential environmental impact.Mutat Res. 2005 Apr 1;571(1-2):221-33. doi: 10.1016/j.mrfmmm.2004.11.017. Epub 2005 Jan 25. Mutat Res. 2005. PMID: 15748649 Review.
-
Molecular mechanisms of DNA damage and repair: progress in plants.Crit Rev Biochem Mol Biol. 2001;36(4):337-97. doi: 10.1080/20014091074219. Crit Rev Biochem Mol Biol. 2001. PMID: 11563486 Review.
-
DNA repair mechanisms in dividing and non-dividing cells.DNA Repair (Amst). 2013 Aug;12(8):620-36. doi: 10.1016/j.dnarep.2013.04.015. Epub 2013 May 16. DNA Repair (Amst). 2013. PMID: 23684800 Free PMC article. Review.
Cited by
-
DNA damage and repair in plants under ultraviolet and ionizing radiations.ScientificWorldJournal. 2015;2015:250158. doi: 10.1155/2015/250158. Epub 2015 Feb 2. ScientificWorldJournal. 2015. PMID: 25729769 Free PMC article. Review.
-
Risk of a second primary cancer after non-melanoma skin cancer in white men and women: a prospective cohort study.PLoS Med. 2013;10(4):e1001433. doi: 10.1371/journal.pmed.1001433. Epub 2013 Apr 23. PLoS Med. 2013. PMID: 23630459 Free PMC article.
-
Identification of Cyclobutane Pyrimidine Dimer-Responsive Genes Using UVB-Irradiated Human Keratinocytes Transfected with In Vitro-Synthesized Photolyase mRNA.PLoS One. 2015 Jun 29;10(6):e0131141. doi: 10.1371/journal.pone.0131141. eCollection 2015. PLoS One. 2015. PMID: 26121660 Free PMC article.
-
The Potential Role of Creatine in Vascular Health.Nutrients. 2021 Mar 5;13(3):857. doi: 10.3390/nu13030857. Nutrients. 2021. PMID: 33807747 Free PMC article. Review.
-
The Role of Senescent Cells in Acquired Drug Resistance and Secondary Cancer in BRAFi-Treated Melanoma.Cancers (Basel). 2021 May 7;13(9):2241. doi: 10.3390/cancers13092241. Cancers (Basel). 2021. PMID: 34066966 Free PMC article. Review.
References
-
- Lubin D, Jensen EH. Effects of clouds and stratospheric ozone depletion on ultraviolet radiation trends. Nature. 1995;377(6551):710–713.
-
- Sinha RP, Kumar HD, Kumar A, Hader DP. Effects of UV-B irradiation on growth, survival, pigmentation and nitrogen metabolism enzymes in cyanobacteria. Acta Protozoologica. 1995;34(3):187–192.
-
- Sinha RP, Rastogi RP, Ambasht NK, Häder D-P. Life of wetland cyanobacteria under enhancing solar UV-B radiation. Proceedings of the National Academy of Sciences, India B. 2008;78:53–65.
-
- Häder D-P, Sinha RP. Solar ultraviolet radiation-induced DNA damage in aquatic organisms: potential environmental impact. Mutation Research. 2005;571(1-2):221–233. - PubMed
-
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC, USA: ASM Press; 2006.
LinkOut - more resources
Full Text Sources
Other Literature Sources
