

RNA sequencing: advances, challenges and opportunities
- PMID: 21191423
- PMCID: PMC3031867
- DOI: 10.1038/nrg2934
RNA sequencing: advances, challenges and opportunities
Abstract
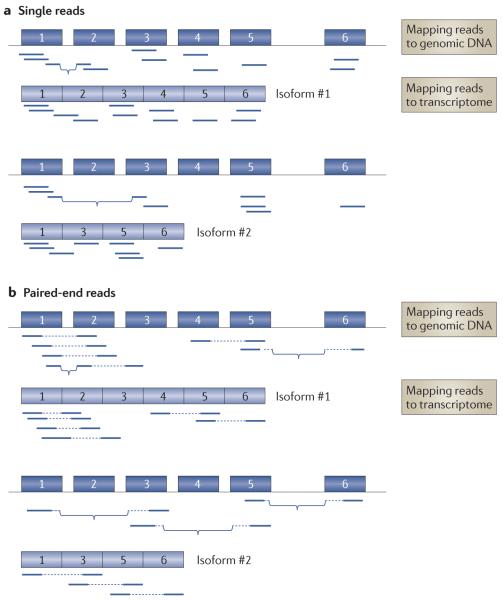
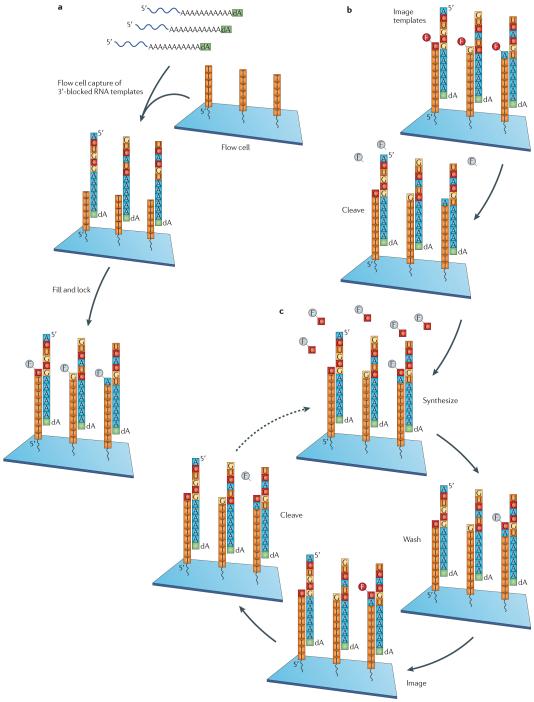
In the few years since its initial application, massively parallel cDNA sequencing, or RNA-seq, has allowed many advances in the characterization and quantification of transcriptomes. Recently, several developments in RNA-seq methods have provided an even more complete characterization of RNA transcripts. These developments include improvements in transcription start site mapping, strand-specific measurements, gene fusion detection, small RNA characterization and detection of alternative splicing events. Ongoing developments promise further advances in the application of RNA-seq, particularly direct RNA sequencing and approaches that allow RNA quantification from very small amounts of cellular materials.
Figures



Similar articles
-
RNA-Seq: a revolutionary tool for transcriptomics.Nat Rev Genet. 2009 Jan;10(1):57-63. doi: 10.1038/nrg2484. Nat Rev Genet. 2009. PMID: 19015660 Free PMC article. Review.
-
Uncovering the complexity of transcriptomes with RNA-Seq.J Biomed Biotechnol. 2010;2010:853916. doi: 10.1155/2010/853916. Epub 2010 Jun 27. J Biomed Biotechnol. 2010. PMID: 20625424 Free PMC article. Review.
-
RAP: RNA-Seq Analysis Pipeline, a new cloud-based NGS web application.BMC Genomics. 2015;16(Suppl 6):S3. doi: 10.1186/1471-2164-16-S6-S3. Epub 2015 Jun 1. BMC Genomics. 2015. PMID: 26046471 Free PMC article.
-
A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling.BMC Genomics. 2010 May 5;11:282. doi: 10.1186/1471-2164-11-282. BMC Genomics. 2010. PMID: 20444259 Free PMC article.
-
Strand-Specific Dual RNA Sequencing of Bronchial Epithelial Cells Infected with Influenza A/H3N2 Viruses Reveals Splicing of Gene Segment 6 and Novel Host-Virus Interactions.J Virol. 2018 Aug 16;92(17):e00518-18. doi: 10.1128/JVI.00518-18. Print 2018 Sep 1. J Virol. 2018. PMID: 29976658 Free PMC article.
Cited by
-
The developmental transcriptome of the mosquito Aedes aegypti, an invasive species and major arbovirus vector.G3 (Bethesda). 2013 Sep 4;3(9):1493-509. doi: 10.1534/g3.113.006742. G3 (Bethesda). 2013. PMID: 23833213 Free PMC article.
-
Comparison of RNA-seq and microarray-based models for clinical endpoint prediction.Genome Biol. 2015 Jun 25;16(1):133. doi: 10.1186/s13059-015-0694-1. Genome Biol. 2015. PMID: 26109056 Free PMC article.
-
Transcriptomic landscape of breast cancers through mRNA sequencing.Sci Rep. 2012;2:264. doi: 10.1038/srep00264. Epub 2012 Feb 14. Sci Rep. 2012. PMID: 22355776 Free PMC article.
-
[Application of RNA sequencing in clinical diagnosis of Mendelian disease].Zhongguo Dang Dai Er Ke Za Zhi. 2020 Oct;22(10):1138-1142. doi: 10.7499/j.issn.1008-8830.2005004. Zhongguo Dang Dai Er Ke Za Zhi. 2020. PMID: 33059815 Free PMC article. Review. Chinese.
-
Recursive Indirect-Paths Modularity (RIP-M) for Detecting Community Structure in RNA-Seq Co-expression Networks.Front Genet. 2016 May 9;7:80. doi: 10.3389/fgene.2016.00080. eCollection 2016. Front Genet. 2016. PMID: 27242890 Free PMC article.
References
-
- Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nature Rev. Genet. 2007;8:413–423. - PubMed
-
-
Metzker ML. Sequencing technologies — the next generation. Nature Rev. Genet. 2010;11:31–46. This Review provides a comprehensive overview of currently available and in-development NGS technologies.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
