

Review
doi: 10.1186/gb-2010-11-12-220.
Epub 2010 Dec 22.
From RNA-seq reads to differential expression results
Affiliations
- PMID: 21176179
- PMCID: PMC3046478
- DOI: 10.1186/gb-2010-11-12-220
Item in Clipboard
Review
From RNA-seq reads to differential expression results
Genome Biol.
2010.
Abstract
Many methods and tools are available for preprocessing high-throughput RNA sequencing data and detecting differential expression.
Figures
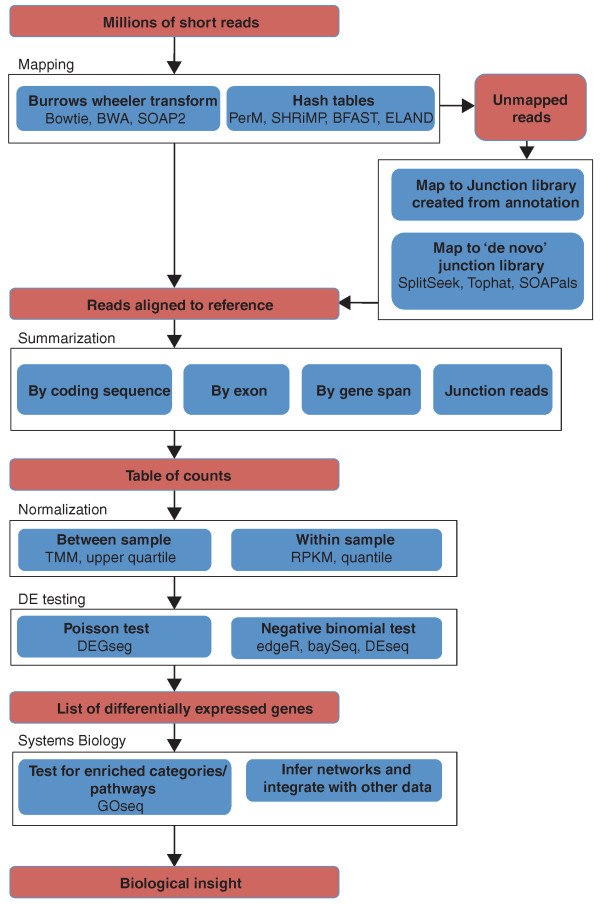
Overview of the RNA-seq analysis pipeline for detecting differential expression. The steps in the pipeline are in red boxes; the methodological components of the pipeline are shown in blue boxes and bold text; software examples and methods for each step (a non-exhaustive list) are shown by regular text in blue boxes. References for the tools and methods shown are listed in Table 1. First, reads are mapped to the reference genome or transcriptome (using junction libraries to map reads that cross exon boundaries); mapped reads are assembled into expression summaries (tables of counts, showing how may reads are in coding region, exon, gene or junction); the data are normalized; statistical testing of differential expression (DE) is performed, producing and a list of genes with associated P-values and fold changes. Systems biology approaches can then be used to gain biological insights from these lists.
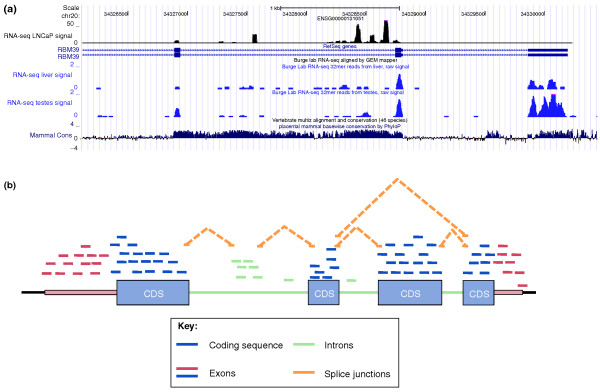
Summarizing mapped reads into a gene level count. (a) Mapped reads from a small region of the RNA-binding protein 39 (RBM39) gene are shown for LNCaP prostate cancer cells [90], human liver and human testis from the UCSC track. The three rows of RNA-seq data (blue and black graphs) are shown as a 'pileup track', where the y-axis at each location measures the number of mapped reads that overlap that location. Also shown are the genomic coordinates, gene model (labeled RBM39; blue boxes indicate exons) and conservation score across vertebrates. It is clear that many reads originate from regions with no known exons. (b) A schematic of a genomic region and reads that might arise from it. Reads are color-coded by the genomic feature from which they originate. Different summarization strategies will result in the inclusion or exclusion of different sets of reads in the table of counts. For example, including only reads coming from known exons will exclude the intronic reads (green) from contributing to the results. Splice junctions are listed as a separate class to emphasize both the potential ambiguity in their assignment (such as which exon should a junction read be assigned to) and the possibility that many of these reads may not be mapped because they are harder to map than continuous reads. CDS, coding sequence.
Similar articles
-
Differential Expression Analysis of RNA-seq Reads: Overview, Taxonomy, and Tools.IEEE/ACM Trans Comput Biol Bioinform. 2020 Mar-Apr;17(2):566-586. doi: 10.1109/TCBB.2018.2873010. Epub 2018 Oct 1. IEEE/ACM Trans Comput Biol Bioinform. 2020. PMID: 30281477 Review.
-
SPARTA: Simple Program for Automated reference-based bacterial RNA-seq Transcriptome Analysis.BMC Bioinformatics. 2016 Feb 4;17:66. doi: 10.1186/s12859-016-0923-y. BMC Bioinformatics. 2016. PMID: 26847232 Free PMC article.
-
Polyester: simulating RNA-seq datasets with differential transcript expression.Bioinformatics. 2015 Sep 1;31(17):2778-84. doi: 10.1093/bioinformatics/btv272. Epub 2015 Apr 28. Bioinformatics. 2015. PMID: 25926345 Free PMC article.
-
In Silico HLA Typing Using Standard RNA-Seq Sequence Reads.Methods Mol Biol. 2015;1310:247-58. doi: 10.1007/978-1-4939-2690-9_20. Methods Mol Biol. 2015. PMID: 26024640
-
Mapping RNA-seq Reads with STAR.Curr Protoc Bioinformatics. 2015 Sep 3;51:11.14.1-11.14.19. doi: 10.1002/0471250953.bi1114s51. Curr Protoc Bioinformatics. 2015. PMID: 26334920 Free PMC article. Review.
Cited by
-
A Survey of MicroRNA Length Variants Contributing to miRNome Complexity in Peach (Prunus Persica L.).Front Plant Sci. 2012 Jul 26;3:165. doi: 10.3389/fpls.2012.00165. eCollection 2012. Front Plant Sci. 2012. PMID: 22855688 Free PMC article.
-
Survey of the Applications of NGS to Whole-Genome Sequencing and Expression Profiling.Genomics Inform. 2012 Mar;10(1):1-8. doi: 10.5808/GI.2012.10.1.1. Epub 2012 Mar 31. Genomics Inform. 2012. PMID: 23105922 Free PMC article.
-
Cardiovascular genomics: a biomarker identification pipeline.IEEE Trans Inf Technol Biomed. 2012 Sep;16(5):809-22. doi: 10.1109/TITB.2012.2199570. Epub 2012 May 16. IEEE Trans Inf Technol Biomed. 2012. PMID: 22614726 Free PMC article. Review.
-
Gene regulation by CcpA and catabolite repression explored by RNA-Seq in Streptococcus mutans.PLoS One. 2013;8(3):e60465. doi: 10.1371/journal.pone.0060465. Epub 2013 Mar 28. PLoS One. 2013. PMID: 23555977 Free PMC article.
-
Targeted DNA and RNA sequencing of fine-needle biopsy FFPE specimens in patients with unresectable hepatocellular carcinoma treated with sorafenib.Oncotarget. 2015 Aug 28;6(25):21636-44. doi: 10.18632/oncotarget.4270. Oncotarget. 2015. PMID: 26046304 Free PMC article.
References
-
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
