

Neuronal pentraxin 1 induction in hypoxic-ischemic neuronal death is regulated via a glycogen synthase kinase-3α/β dependent mechanism
- PMID: 21130869
- PMCID: PMC3056287
- DOI: 10.1016/j.cellsig.2010.11.021
Neuronal pentraxin 1 induction in hypoxic-ischemic neuronal death is regulated via a glycogen synthase kinase-3α/β dependent mechanism
Abstract
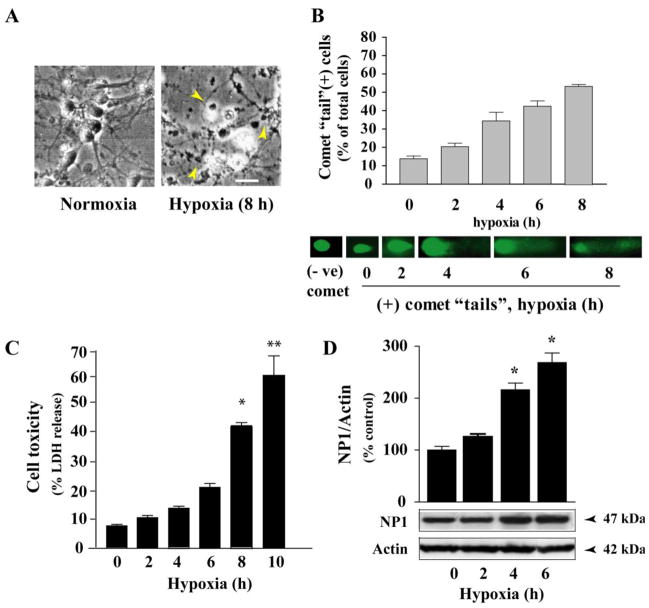
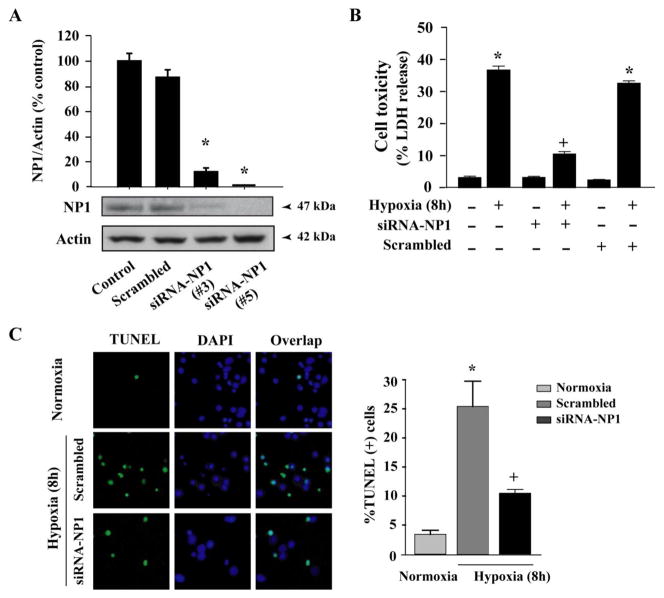
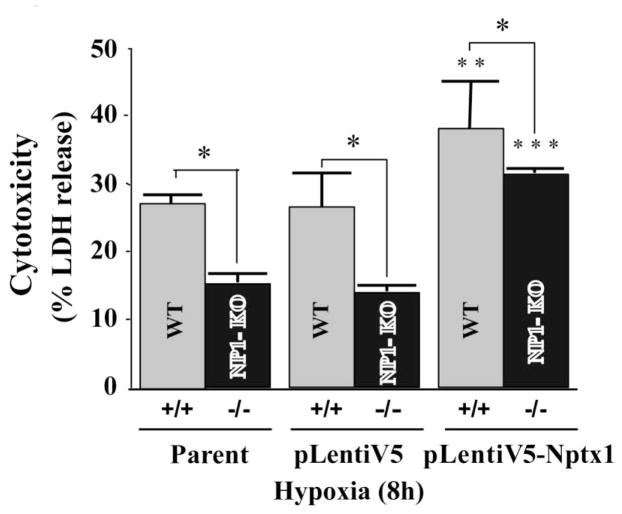
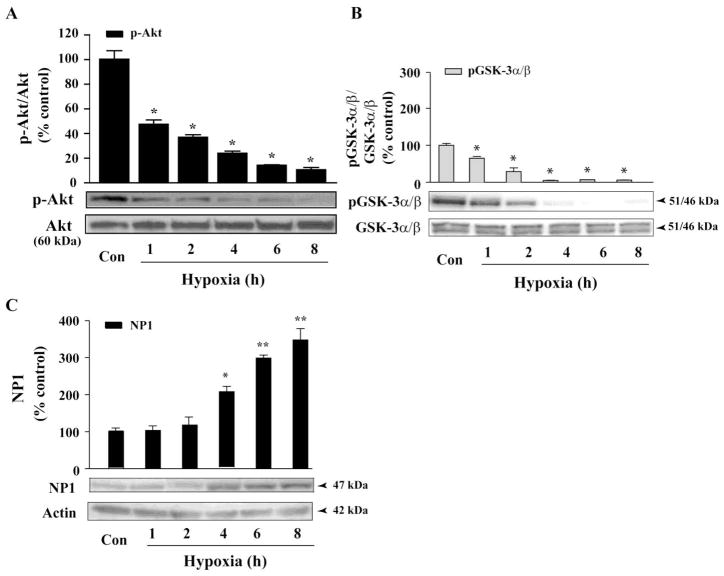
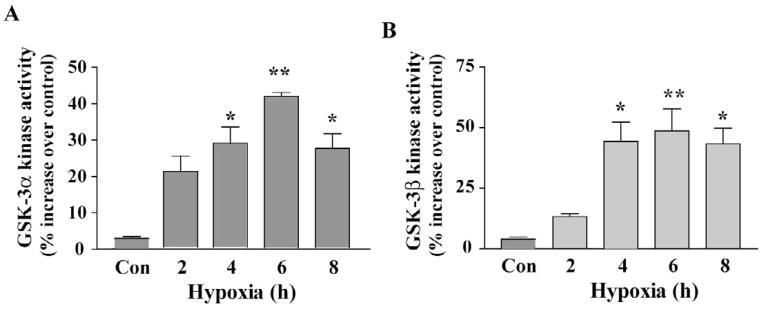
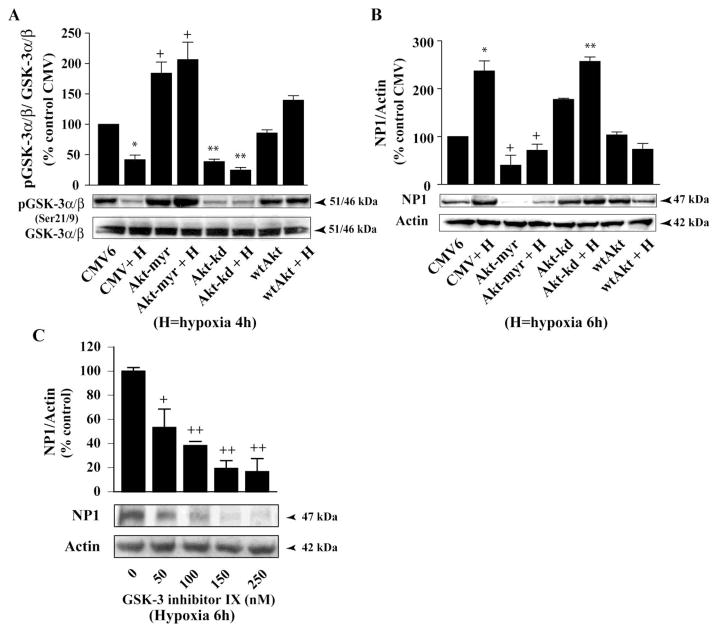
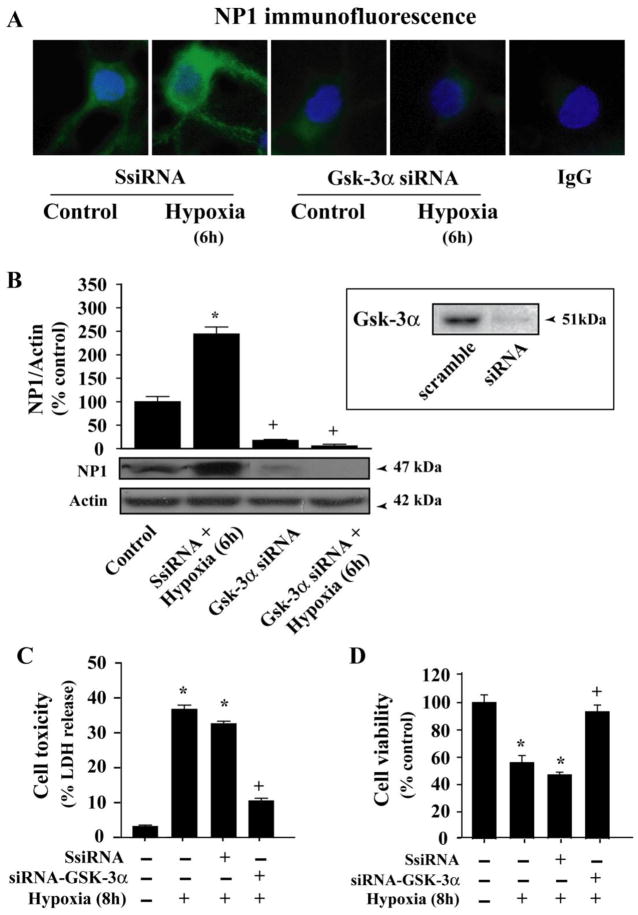
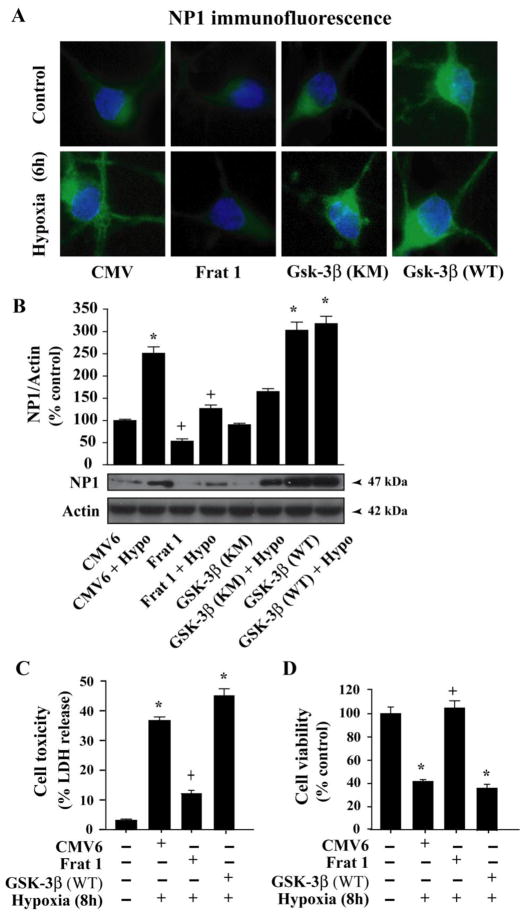
Intracellular signaling pathways that regulate the production of lethal proteins in central neurons are not fully characterized. Previously, we reported induction of a novel neuronal protein neuronal pentraxin 1 (NP1) in neonatal brain injury following hypoxia-ischemia (HI); however, how NP1 is induced in hypoxic-ischemic neuronal death remains elusive. Here, we have elucidated the intracellular signaling regulation of NP1 induction in neuronal death. Primary cortical neurons showed a hypoxic-ischemia time-dependent increase in cell death and that NP1 induction preceded the actual neuronal death. NP1 gene silencing by NP1-specific siRNA significantly reduced neuronal death. The specificity of NP1 induction in neuronal death was further confirmed by using NP1 (-/-) null primary cortical neurons. Declines in phospho-Akt (i.e. deactivation) were observed concurrent with decreased phosphorylation of its downstream substrate GSK-3α/β (at Ser21/Ser9) (i.e. activation) and increased GSK-3α and GSK-3β kinase activities, which occurred prior to NP1 induction. Expression of a dominant-negative inhibitor of Akt (Akt-kd) blocked phosphorylation of GSK-3α/β and subsequently enhanced NP1 induction. Whereas, overexpression of constitutively activated Akt (Akt-myr) or wild-type Akt (wtAkt) increased GSK-α/β phosphorylation and attenuated NP1 induction. Transfection of neurons with GSK-3α siRNA completely blocked NP1 induction and cell death. Similarly, overexpression of the GSK-3β inhibitor Frat1 or the kinase mutant GSK-3βKM, but not the wild-type GSK-3βWT, blocked NP1 induction and rescued neurons from death. Our findings clearly implicate both GSK-3α- and GSK-3β-dependent mechanism of NP1 induction and point to a novel mechanism in the regulation of hypoxic-ischemic neuronal death.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Critical role of neuronal pentraxin 1 in mitochondria-mediated hypoxic-ischemic neuronal injury.Neurobiol Dis. 2013 Feb;50:59-68. doi: 10.1016/j.nbd.2012.10.003. Epub 2012 Oct 12. Neurobiol Dis. 2013. PMID: 23069675 Free PMC article.
-
Genetic deletion of neuronal pentraxin 1 expression prevents brain injury in a neonatal mouse model of cerebral hypoxia-ischemia.Neurobiol Dis. 2015 Mar;75:15-30. doi: 10.1016/j.nbd.2014.12.016. Epub 2014 Dec 29. Neurobiol Dis. 2015. PMID: 25554688 Free PMC article.
-
Neuronal pentraxin 1: a novel mediator of hypoxic-ischemic injury in neonatal brain.J Neurosci. 2004 Apr 28;24(17):4187-96. doi: 10.1523/JNEUROSCI.0347-04.2004. J Neurosci. 2004. PMID: 15115814 Free PMC article.
-
Hypoxic-ischemic injury in neonatal brain: involvement of a novel neuronal molecule in neuronal cell death and potential target for neuroprotection.Int J Dev Neurosci. 2008 Feb;26(1):93-101. doi: 10.1016/j.ijdevneu.2007.08.013. Epub 2007 Sep 7. Int J Dev Neurosci. 2008. PMID: 17936538 Free PMC article. Review.
-
Role of GSK-3 in Cardiac Health: Focusing on Cardiac Remodeling and Heart Failure.Curr Drug Targets. 2021;22(13):1568-1576. doi: 10.2174/1389450122666210224105430. Curr Drug Targets. 2021. PMID: 33655828 Review.
Cited by
-
Neuronal pentraxin 1: A synaptic-derived plasma biomarker in Alzheimer's disease.Neurobiol Dis. 2018 Jun;114:120-128. doi: 10.1016/j.nbd.2018.02.014. Epub 2018 Mar 6. Neurobiol Dis. 2018. PMID: 29501530 Free PMC article.
-
GSK-3β Contributes to Parkinsonian Dopaminergic Neuron Death: Evidence From Conditional Knockout Mice and Tideglusib.Front Mol Neurosci. 2020 Jun 3;13:81. doi: 10.3389/fnmol.2020.00081. eCollection 2020. Front Mol Neurosci. 2020. PMID: 32581704 Free PMC article.
-
As a downstream target of the AKT pathway, NPTX1 inhibits proliferation and promotes apoptosis in hepatocellular carcinoma.Biosci Rep. 2019 Jun 4;39(6):BSR20181662. doi: 10.1042/BSR20181662. Print 2019 Jun 28. Biosci Rep. 2019. PMID: 31113871 Free PMC article.
-
Treatment advances in neonatal neuroprotection and neurointensive care.Lancet Neurol. 2011 Apr;10(4):372-82. doi: 10.1016/S1474-4422(11)70016-3. Lancet Neurol. 2011. PMID: 21435600 Free PMC article. Review.
-
G-CSF ameliorates neuronal apoptosis through GSK-3β inhibition in neonatal hypoxia-ischemia in rats.Exp Neurol. 2015 Jan;263:141-9. doi: 10.1016/j.expneurol.2014.10.004. Epub 2014 Oct 18. Exp Neurol. 2015. PMID: 25448005 Free PMC article.
References
-
- Lorenz JM, Wooliever DE, Jetton JR, Paneth N. Arch Pediatr Adolesc Med. 1998;152(5):425–435. - PubMed
-
- Northington FJ, Ferriero DM, Martin LJ. Dev Neurosci. 2001;23(3):186–191. - PubMed
-
- Johnston MV. Brain Dev. 1997;19(4):235–239. - PubMed
-
- Volpe JJ. Ment Retard Dev Disabil Res Rev. 2001;7(1):56–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
