

Scalable rule-based modelling of allosteric proteins and biochemical networks
- PMID: 21079669
- PMCID: PMC2973810
- DOI: 10.1371/journal.pcbi.1000975
Scalable rule-based modelling of allosteric proteins and biochemical networks
Abstract
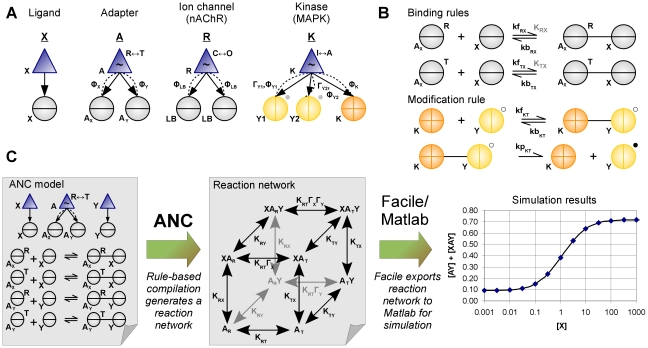
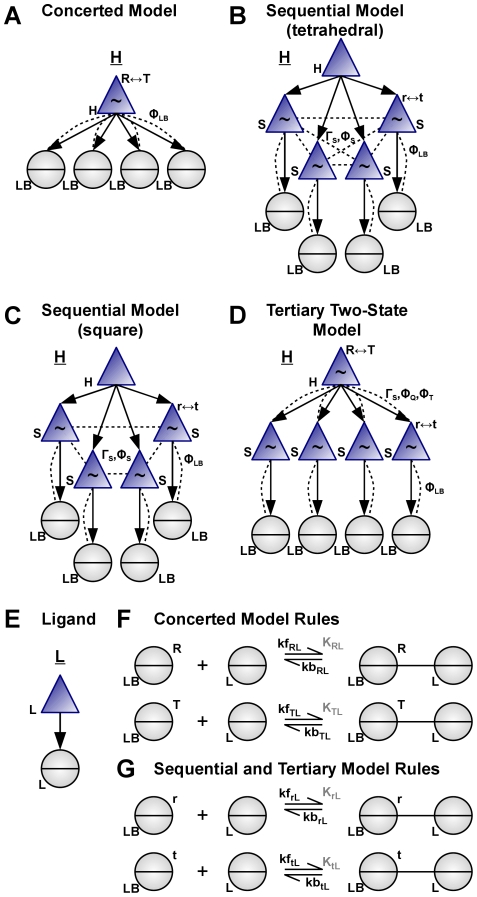
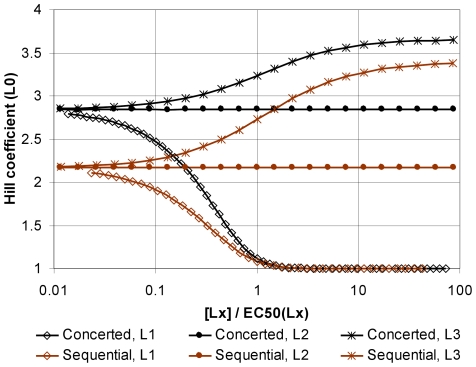
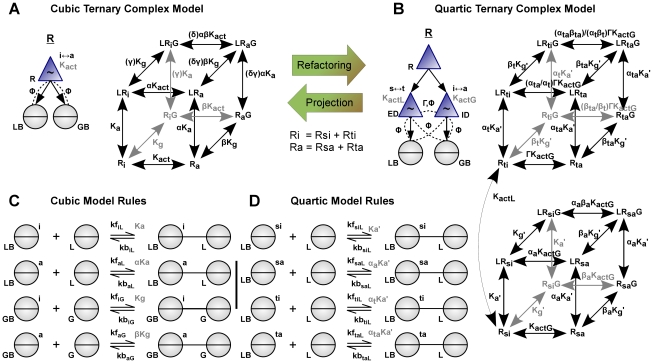
Much of the complexity of biochemical networks comes from the information-processing abilities of allosteric proteins, be they receptors, ion-channels, signalling molecules or transcription factors. An allosteric protein can be uniquely regulated by each combination of input molecules that it binds. This "regulatory complexity" causes a combinatorial increase in the number of parameters required to fit experimental data as the number of protein interactions increases. It therefore challenges the creation, updating, and re-use of biochemical models. Here, we propose a rule-based modelling framework that exploits the intrinsic modularity of protein structure to address regulatory complexity. Rather than treating proteins as "black boxes", we model their hierarchical structure and, as conformational changes, internal dynamics. By modelling the regulation of allosteric proteins through these conformational changes, we often decrease the number of parameters required to fit data, and so reduce over-fitting and improve the predictive power of a model. Our method is thermodynamically grounded, imposes detailed balance, and also includes molecular cross-talk and the background activity of enzymes. We use our Allosteric Network Compiler to examine how allostery can facilitate macromolecular assembly and how competitive ligands can change the observed cooperativity of an allosteric protein. We also develop a parsimonious model of G protein-coupled receptors that explains functional selectivity and can predict the rank order of potency of agonists acting through a receptor. Our methodology should provide a basis for scalable, modular and executable modelling of biochemical networks in systems and synthetic biology.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
[G protein-coupled receptors: allosteric regulators of cell metabolism].Med Sci (Paris). 2012 Oct;28(10):852-7. doi: 10.1051/medsci/20122810013. Epub 2012 Oct 12. Med Sci (Paris). 2012. PMID: 23067416 Review. French.
-
Allostery in Its Many Disguises: From Theory to Applications.Structure. 2019 Apr 2;27(4):566-578. doi: 10.1016/j.str.2019.01.003. Epub 2019 Feb 7. Structure. 2019. PMID: 30744993 Free PMC article. Review.
-
Symmetry, Rigidity, and Allosteric Signaling: From Monomeric Proteins to Molecular Machines.Chem Rev. 2019 Jun 26;119(12):6788-6821. doi: 10.1021/acs.chemrev.8b00760. Epub 2019 Apr 24. Chem Rev. 2019. PMID: 31017391
-
Allosteric communication and signal transduction in proteins.Curr Opin Struct Biol. 2024 Feb;84:102737. doi: 10.1016/j.sbi.2023.102737. Epub 2024 Jan 3. Curr Opin Struct Biol. 2024. PMID: 38171189 Review.
-
Conformational spread: the propagation of allosteric states in large multiprotein complexes.Annu Rev Biophys Biomol Struct. 2004;33:53-73. doi: 10.1146/annurev.biophys.33.110502.132703. Annu Rev Biophys Biomol Struct. 2004. PMID: 15139804 Review.
Cited by
-
Derivative-Free Optimization of Rate Parameters of Capsid Assembly Models from Bulk in Vitro Data.IEEE/ACM Trans Comput Biol Bioinform. 2017 Jul-Aug;14(4):844-855. doi: 10.1109/TCBB.2016.2563421. Epub 2016 May 5. IEEE/ACM Trans Comput Biol Bioinform. 2017. PMID: 27168601 Free PMC article.
-
Bivalent Ligands for Protein Degradation in Drug Discovery.Comput Struct Biotechnol J. 2019 Jan 25;17:160-176. doi: 10.1016/j.csbj.2019.01.006. eCollection 2019. Comput Struct Biotechnol J. 2019. PMID: 30788082 Free PMC article. Review.
-
BioJazz: in silico evolution of cellular networks with unbounded complexity using rule-based modeling.Nucleic Acids Res. 2015 Oct 30;43(19):e123. doi: 10.1093/nar/gkv595. Epub 2015 Jun 22. Nucleic Acids Res. 2015. PMID: 26101250 Free PMC article.
-
Retour aux sources: defining the structural basis of glutamate receptor activation.J Physiol. 2015 Jan 1;593(1):97-110. doi: 10.1113/jphysiol.2014.277921. Epub 2014 Oct 21. J Physiol. 2015. PMID: 25556791 Free PMC article. Review.
-
Modeling for (physical) biologists: an introduction to the rule-based approach.Phys Biol. 2015 Jul 16;12(4):045007. doi: 10.1088/1478-3975/12/4/045007. Phys Biol. 2015. PMID: 26178138 Free PMC article. Review.
References
-
- Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–52. - PubMed
-
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. - PubMed
-
- Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. - PubMed
-
- Bray D. Protein molecules as computational elements in living cells. Nature. 1995;376:307–312. - PubMed
-
- Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
