

IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1
- PMID: 21072055
- PMCID: PMC3131923
- DOI: 10.1038/cdd.2010.143
IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1
Abstract
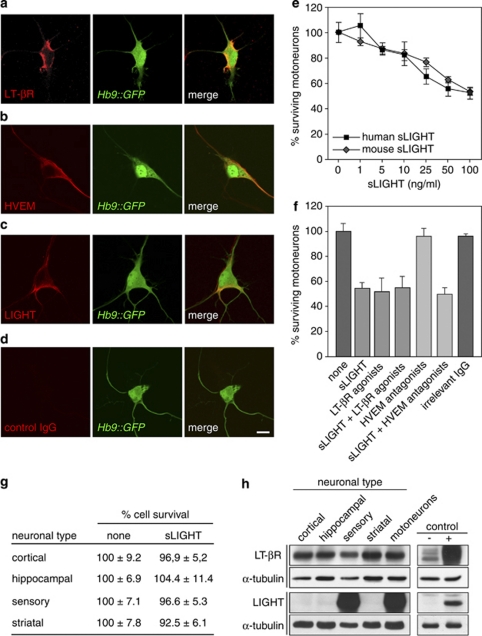
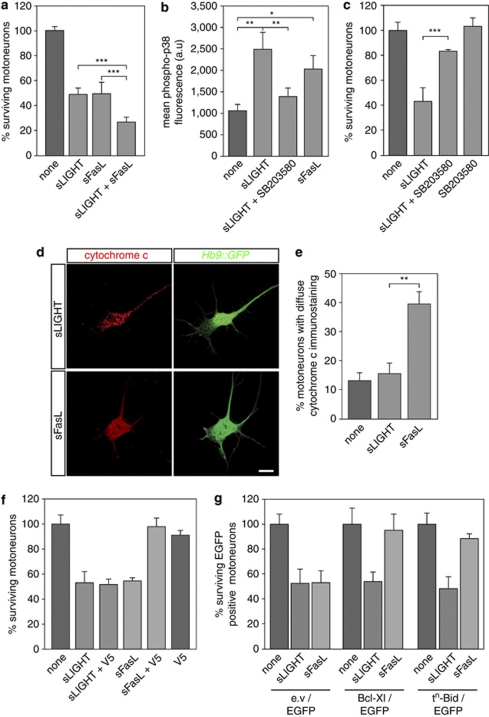
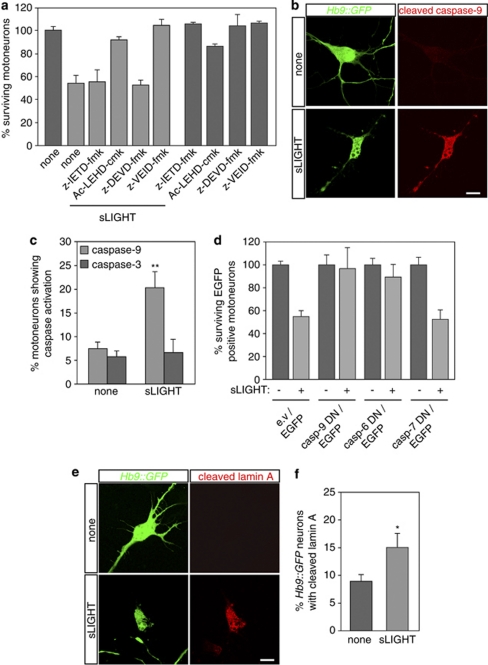
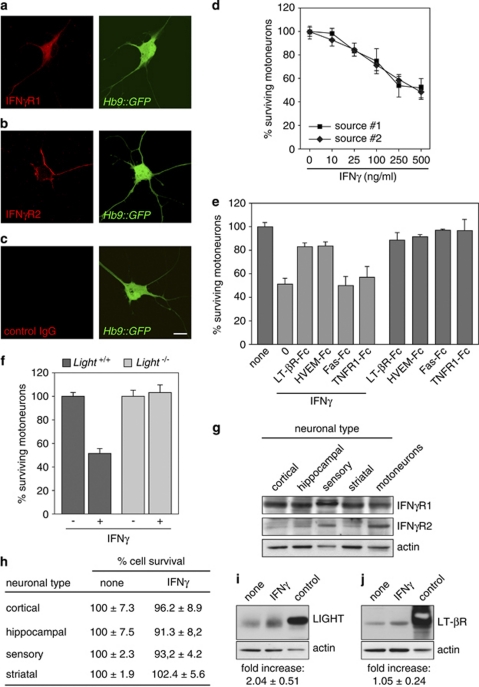
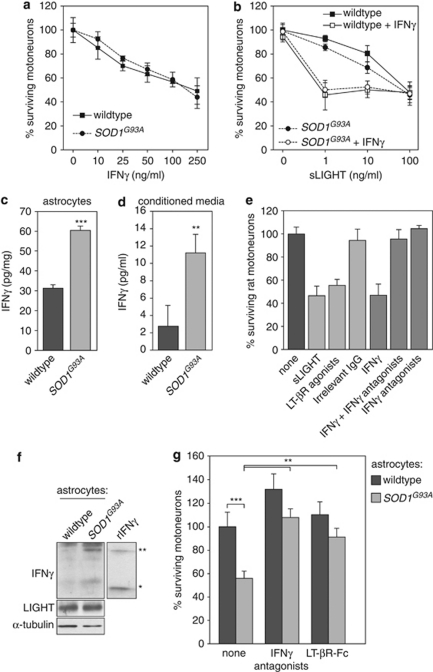
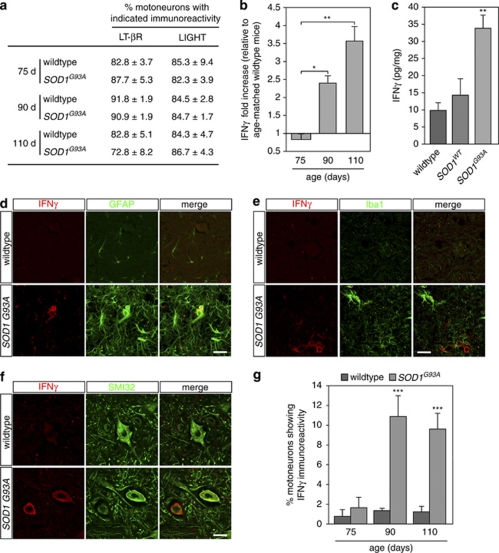
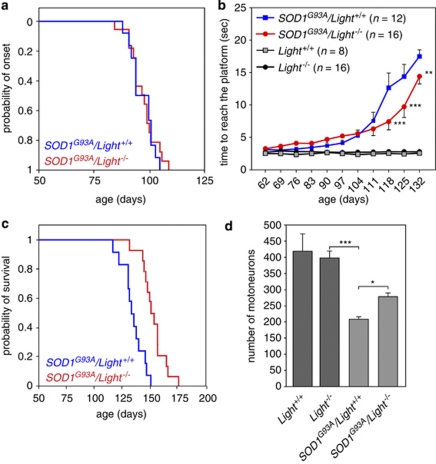
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disease that primarily affects motoneurons in the brain and spinal cord. Dominant mutations in superoxide dismutase-1 (SOD1) cause a familial form of ALS. Mutant SOD1-damaged glial cells contribute to ALS pathogenesis by releasing neurotoxic factors, but the mechanistic basis of the motoneuron-specific elimination is poorly understood. Here, we describe a motoneuron-selective death pathway triggered by activation of lymphotoxin-β receptor (LT-βR) by LIGHT, and operating by a novel signaling scheme. We show that astrocytes expressing mutant SOD1 mediate the selective death of motoneurons through the proinflammatory cytokine interferon-γ (IFNγ), which activates the LIGHT-LT-βR death pathway. The expression of LIGHT and LT-βR by motoneurons in vivo correlates with the preferential expression of IFNγ by motoneurons and astrocytes at disease onset and symptomatic stage in ALS mice. Importantly, the genetic ablation of Light in an ALS mouse model retards progression, but not onset, of the disease and increases lifespan. We propose that IFNγ contributes to a cross-talk between motoneurons and astrocytes causing the selective loss of some motoneurons following activation of the LIGHT-induced death pathway.
Figures

Similar articles
-
Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis.Brain. 2011 Sep;134(Pt 9):2627-41. doi: 10.1093/brain/awr193. Brain. 2011. PMID: 21908873 Free PMC article.
-
Elevated levels of IFNγ and LIGHT in the spinal cord of patients with sporadic amyotrophic lateral sclerosis.Eur J Neurol. 2012 May;19(5):752-9, e45-6. doi: 10.1111/j.1468-1331.2011.03623.x. Epub 2012 Jan 4. Eur J Neurol. 2012. PMID: 22221541
-
Cytotoxic CD8+ T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons.Proc Natl Acad Sci U S A. 2019 Feb 5;116(6):2312-2317. doi: 10.1073/pnas.1815961116. Epub 2019 Jan 23. Proc Natl Acad Sci U S A. 2019. PMID: 30674678 Free PMC article.
-
Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis.Mol Neurobiol. 2012 Feb;45(1):30-42. doi: 10.1007/s12035-011-8217-x. Epub 2011 Nov 10. Mol Neurobiol. 2012. PMID: 22072396 Free PMC article. Review.
-
[Proximal axonal injuries as experimental models for adult motoneuron degeneration].Brain Nerve. 2007 Oct;59(10):1179-86. Brain Nerve. 2007. PMID: 17969359 Review. Japanese.
Cited by
-
Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS.J Neurosci. 2012 Apr 4;32(14):4901-12. doi: 10.1523/JNEUROSCI.5431-11.2012. J Neurosci. 2012. PMID: 22492046 Free PMC article.
-
Astrocyte-targeting RNA interference against mutated superoxide dismutase 1 induces motoneuron plasticity and protects fast-fatigable motor units in a mouse model of amyotrophic lateral sclerosis.Glia. 2022 May;70(5):842-857. doi: 10.1002/glia.24140. Epub 2022 Jan 3. Glia. 2022. PMID: 34978340 Free PMC article.
-
Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis.Curr Pharm Des. 2017;23(33):5010-5021. doi: 10.2174/1381612823666170622095802. Curr Pharm Des. 2017. PMID: 28641533 Free PMC article. Review.
-
Astrocyte-Neuron Interactions Contributing to Amyotrophic Lateral Sclerosis Progression.Adv Neurobiol. 2024;39:285-318. doi: 10.1007/978-3-031-64839-7_12. Adv Neurobiol. 2024. PMID: 39190080 Review.
-
Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis.J Biol Chem. 2013 May 31;288(22):15699-711. doi: 10.1074/jbc.M112.425066. Epub 2013 Apr 16. J Biol Chem. 2013. PMID: 23592792 Free PMC article.
References
-
- Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. 2010;33:409–440. - PubMed
-
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. - PubMed
-
- Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97:687–696. - PubMed
-
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
