

A panel of human cell-based artificial APC enables the expansion of long-lived antigen-specific CD4+ T cells restricted by prevalent HLA-DR alleles
- PMID: 21059769
- PMCID: PMC2994545
- DOI: 10.1093/intimm/dxq440
A panel of human cell-based artificial APC enables the expansion of long-lived antigen-specific CD4+ T cells restricted by prevalent HLA-DR alleles
Abstract
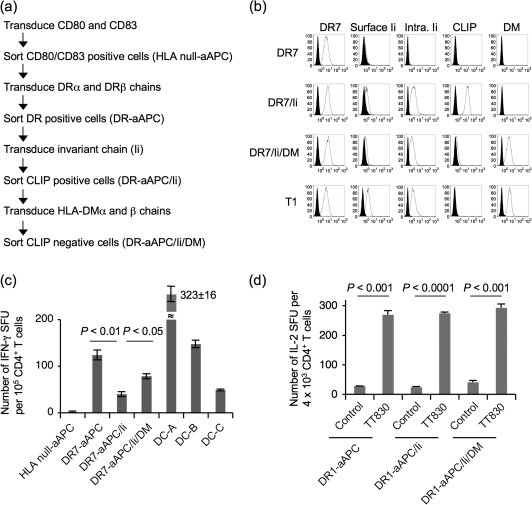
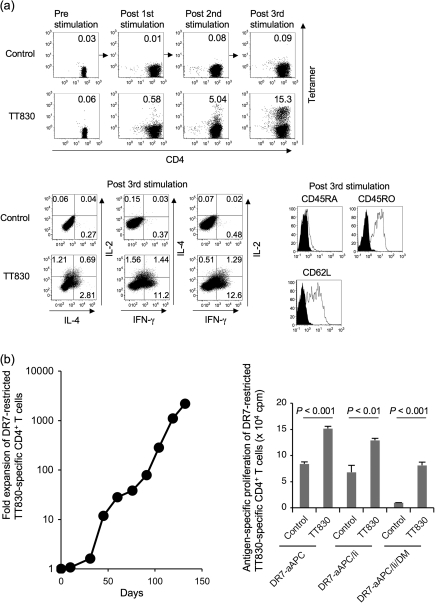
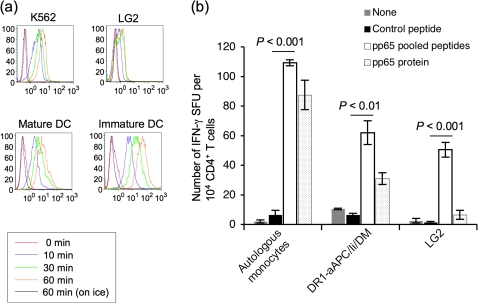
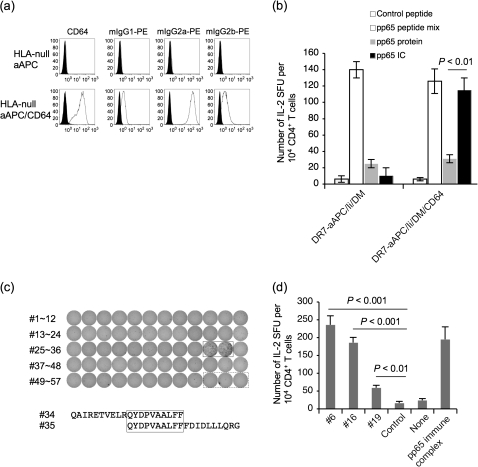
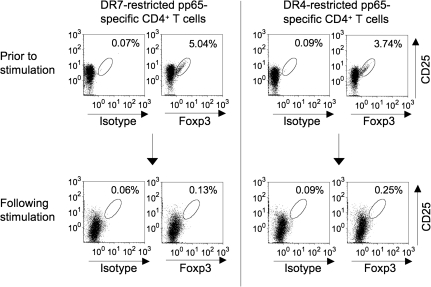
Many preclinical experiments have attested to the critical role of CD4(+) T cell help in CD8(+) cytotoxic T lymphocyte (CTL)-mediated immunity. Recent clinical trials have demonstrated that reinfusion of CD4(+) T cells can induce responses in infectious diseases and cancer. However, few standardized and versatile systems exist to expand antigen-specific CD4(+) T(h) for clinical use. K562 is a human erythroleukemic cell line, which lacks expression of HLA class I and class II, invariant chain and HLA-DM but expresses adhesion molecules such as intercellular adhesion molecule-1 and leukocyte function-associated antigen-3. With this unique immunologic phenotype, K562 has been tested in clinical trials of cancer immunotherapy. Previously, we created a K562-based artificial antigen-presenting cell (aAPC) that generates ex vivo long-lived HLA-A2-restricted CD8(+) CTL with a central/effector memory phenotype armed with potent effector function. We successfully generated a clinical version of this aAPC and conducted a clinical trial where large numbers of anti-tumor CTL are reinfused to cancer patients. In this article, we shifted focus to CD4(+) T cells and developed a panel of novel K562-derived aAPC, where each expresses a different single HLA-DR allele, invariant chain, HLA-DM, CD80, CD83 and CD64; takes up soluble protein by endocytosis and processes and presents CD4(+) T-cell peptides. Using this aAPC, we were able to determine novel DR-restricted CD4(+) T-cell epitopes and expand long-lived CD4(+) T-cells specific for multiple antigens without growing bystander Foxp3(+) regulatory T cells. Our results suggest that K562-based aAPC may serve as a translatable platform to generate both antigen-specific CD8(+) CTL and CD4(+) T(h).
Figures
Similar articles
-
Induction of HLA-DP4-restricted anti-survivin Th1 and Th2 responses using an artificial antigen-presenting cell.Clin Cancer Res. 2011 Aug 15;17(16):5392-401. doi: 10.1158/1078-0432.CCR-10-3083. Epub 2011 Jun 24. Clin Cancer Res. 2011. PMID: 21705450 Free PMC article.
-
Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell.Clin Cancer Res. 2007 Mar 15;13(6):1857-67. doi: 10.1158/1078-0432.CCR-06-1905. Clin Cancer Res. 2007. PMID: 17363542
-
Comprehensive Analysis of CD4+ T Cell Responses to CMV pp65 Antigen Restricted by Single HLA-DR, -DQ, and -DP Allotype Within an Individual.Front Immunol. 2021 Feb 15;11:602014. doi: 10.3389/fimmu.2020.602014. eCollection 2020. Front Immunol. 2021. PMID: 33658991 Free PMC article.
-
HLA-Class II Artificial Antigen Presenting Cells in CD4+ T Cell-Based Immunotherapy.Front Immunol. 2019 May 17;10:1081. doi: 10.3389/fimmu.2019.01081. eCollection 2019. Front Immunol. 2019. PMID: 31156634 Free PMC article. Review.
-
Overview of a HLA-Ig based "Lego-like system" for T cell monitoring, modulation and expansion.Immunol Res. 2010 Jul;47(1-3):248-56. doi: 10.1007/s12026-009-8156-z. Immunol Res. 2010. PMID: 20087680 Free PMC article. Review.
Cited by
-
Adoptive T Cell Therapy Targeting CD1 and MR1.Front Immunol. 2015 May 20;6:247. doi: 10.3389/fimmu.2015.00247. eCollection 2015. Front Immunol. 2015. PMID: 26052329 Free PMC article. Review.
-
Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy.Oncoimmunology. 2012 Sep 1;1(6):908-916. doi: 10.4161/onci.21205. Oncoimmunology. 2012. PMID: 23162758 Free PMC article.
-
Mouse and Human CD1d-Self-Lipid Complexes Are Recognized Differently by Murine Invariant Natural Killer T Cell Receptors.PLoS One. 2016 May 23;11(5):e0156114. doi: 10.1371/journal.pone.0156114. eCollection 2016. PLoS One. 2016. PMID: 27213277 Free PMC article.
-
Human cell-based artificial antigen-presenting cells for cancer immunotherapy.Immunol Rev. 2014 Jan;257(1):191-209. doi: 10.1111/imr.12129. Immunol Rev. 2014. PMID: 24329798 Free PMC article. Review.
-
Artificial antigen-presenting cells expressing AFP(158-166) peptide and interleukin-15 activate AFP-specific cytotoxic T lymphocytes.Oncotarget. 2016 Apr 5;7(14):17579-90. doi: 10.18632/oncotarget.8198. Oncotarget. 2016. PMID: 27007051 Free PMC article.
References
-
- Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu. Rev. Immunol. 2007;25:243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
