

A common molecular mechanism underlies two phenotypically distinct 17p13.1 microdeletion syndromes
- PMID: 21056402
- PMCID: PMC2978979
- DOI: 10.1016/j.ajhg.2010.10.007
A common molecular mechanism underlies two phenotypically distinct 17p13.1 microdeletion syndromes
Abstract
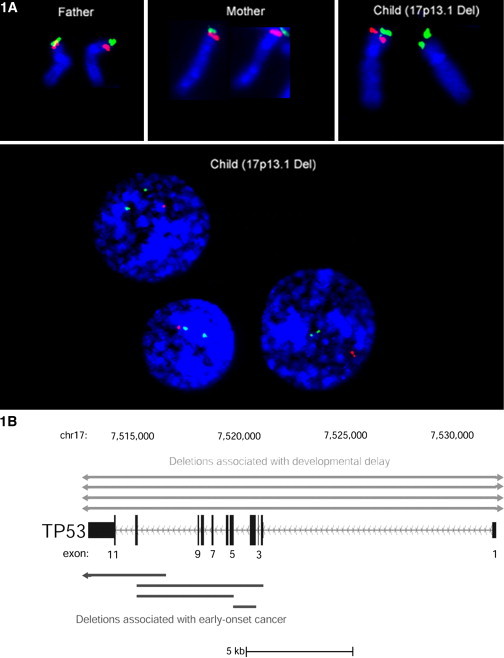
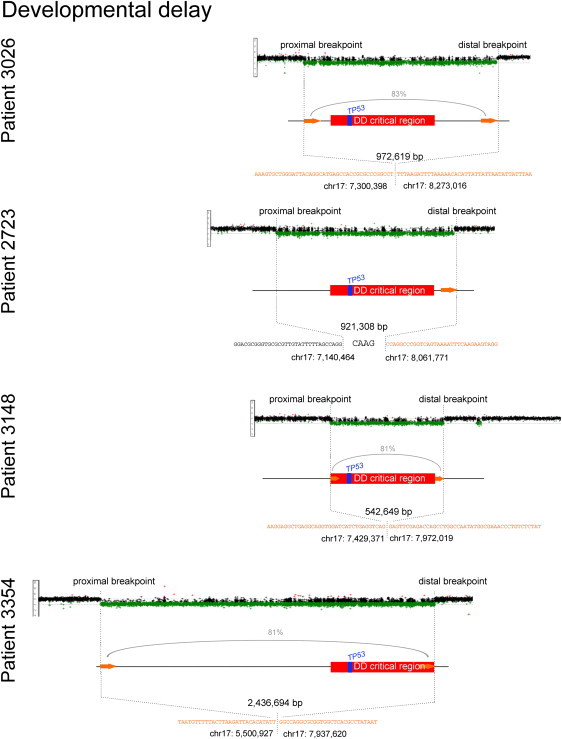
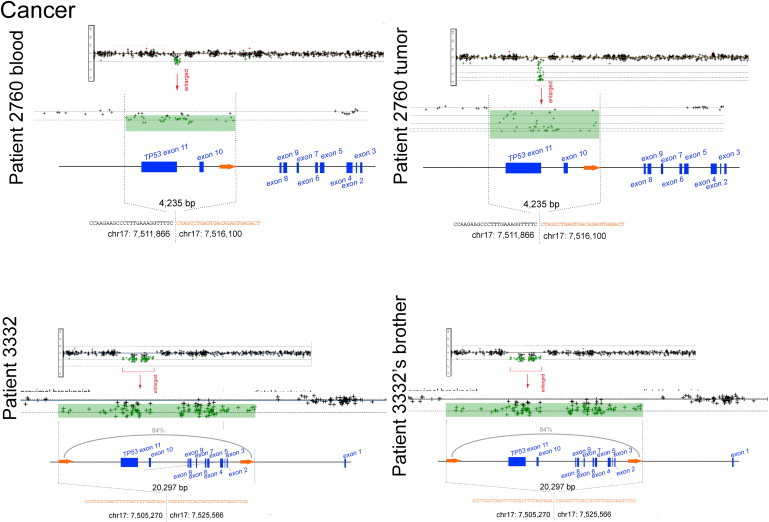
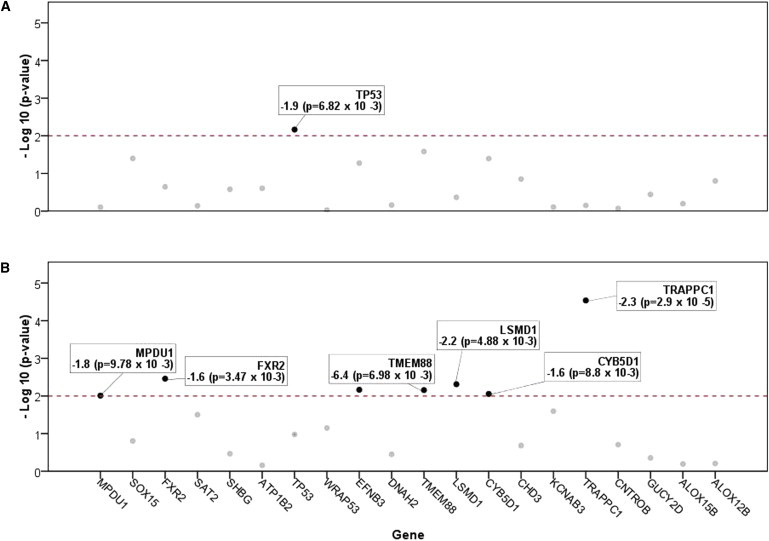
DNA copy-number variations (CNVs) underlie many neuropsychiatric conditions, but they have been less studied in cancer. We report the association of a 17p13.1 CNV, childhood-onset developmental delay (DD), and cancer. Through a screen of over 4000 patients with diverse diagnoses, we identified eight probands harboring microdeletions at TP53 (17p13.1). We used a purpose-built high-resolution array with 93.75% breakpoint accuracy to fine map these microdeletions. Four patients were found to have a common phenotype including DD, hypotonia, and hand and foot abnormalities, constituting a unique syndrome. Notably, these patients were not affected with cancer. Moreover, none of the TP53-deletion patients affected with cancer (n = 4) had neurocognitive impairments. DD patients have larger deletions, which encompass but do not disrupt TP53, whereas cancer-affected patients harbor CNVs with at least one breakpoint within TP53. Most 17p13.1 deletions arise by Alu-mediated nonallelic homologous recombination. Furthermore, we identify a critical genomic region associated with DD and containing six underexpressed genes. We conclude that, although they overlap, 17p13.1 CNVs are associated with distinct phenotypes depending on the position of the breakpoint with respect to TP53. Further, detailed characterization of breakpoints revealed a common formation signature. Future studies should consider whether other loci in the genome also give rise to phenotypically distinct disorders by means of a common mechanism, resulting in a similar formation signature.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Pseudoautosomal abnormalities in terminal AZFb+c deletions are associated with isochromosomes Yp and may lead to abnormal growth and neuropsychiatric function.Hum Reprod. 2017 Feb;32(2):465-475. doi: 10.1093/humrep/dew333. Epub 2017 Jan 5. Hum Reprod. 2017. PMID: 28057878
-
Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes.J Med Genet. 2010 May;47(5):299-311. doi: 10.1136/jmg.2009.069906. J Med Genet. 2010. PMID: 20452996
-
A de novo microtriplication at 4q21.21-q21.22 in a patient with a vascular malignant hemangioma, elongated sigmoid colon, developmental delay, and absence of speech.Am J Med Genet A. 2016 Aug;170(8):2089-96. doi: 10.1002/ajmg.a.37754. Epub 2016 Jun 10. Am J Med Genet A. 2016. PMID: 27288323
-
Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3.Front Genet. 2018 Mar 23;9:80. doi: 10.3389/fgene.2018.00080. eCollection 2018. Front Genet. 2018. PMID: 29628935 Free PMC article. Review.
-
Origins and breakpoint analyses of copy number variations: up close and personal.Cytogenet Genome Res. 2011;135(3-4):271-6. doi: 10.1159/000330267. Epub 2011 Aug 12. Cytogenet Genome Res. 2011. PMID: 21846967 Review.
Cited by
-
Dissecting the complexity of CNV pathogenicity: insights from Drosophila and zebrafish models.Curr Opin Genet Dev. 2021 Jun;68:79-87. doi: 10.1016/j.gde.2021.02.013. Epub 2021 Mar 31. Curr Opin Genet Dev. 2021. PMID: 33812298 Free PMC article. Review.
-
Germline DNA copy number variation in familial and early-onset breast cancer.Breast Cancer Res. 2012 Feb 7;14(1):R24. doi: 10.1186/bcr3109. Breast Cancer Res. 2012. PMID: 22314128 Free PMC article.
-
Age-dependent copy number variations of TP53 tumour suppressor gene associated with altered phosphorylation status of p53 protein in sporadic schwannomas.J Neurooncol. 2019 Jul;143(3):369-379. doi: 10.1007/s11060-019-03176-1. Epub 2019 May 2. J Neurooncol. 2019. PMID: 31049827
-
Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome.Am J Hum Genet. 2016 Aug 4;99(2):318-36. doi: 10.1016/j.ajhg.2015.04.023. Am J Hum Genet. 2016. PMID: 27486776 Free PMC article.
-
On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease.Hum Mutat. 2011 Oct;32(10):1075-99. doi: 10.1002/humu.21557. Epub 2011 Sep 2. Hum Mutat. 2011. PMID: 21853507 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
