

The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x
- PMID: 20980256
- PMCID: PMC3012990
- DOI: 10.1074/jbc.M110.162644
The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x
Abstract
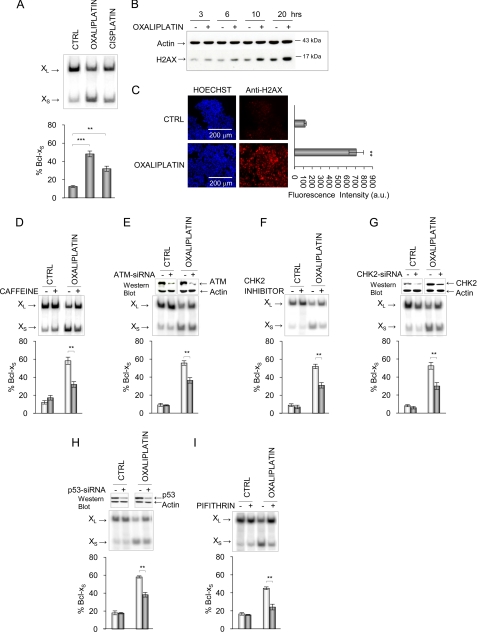
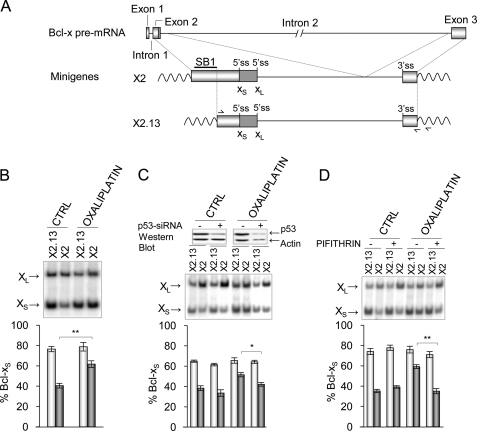
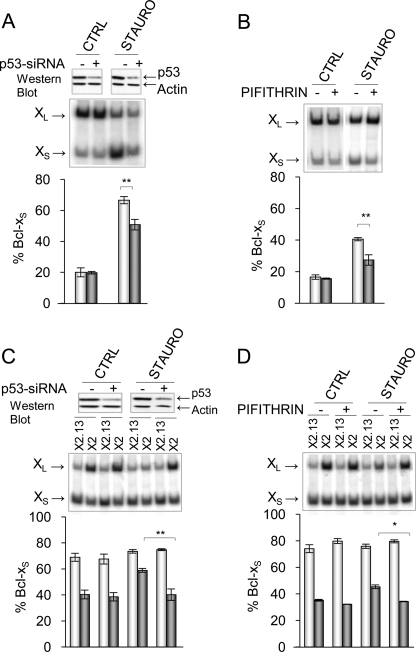
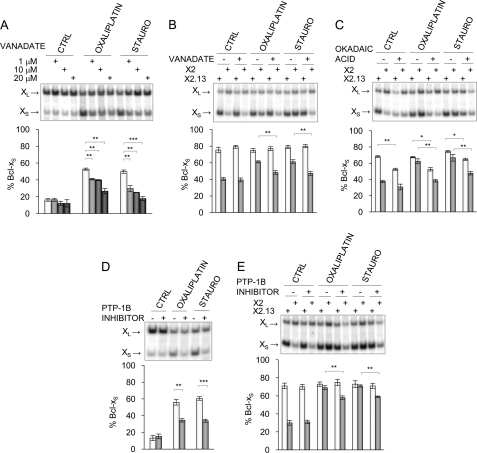
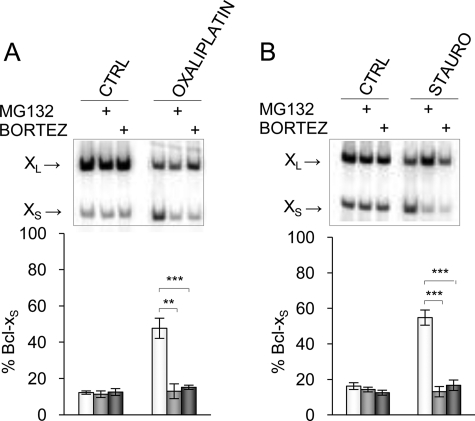
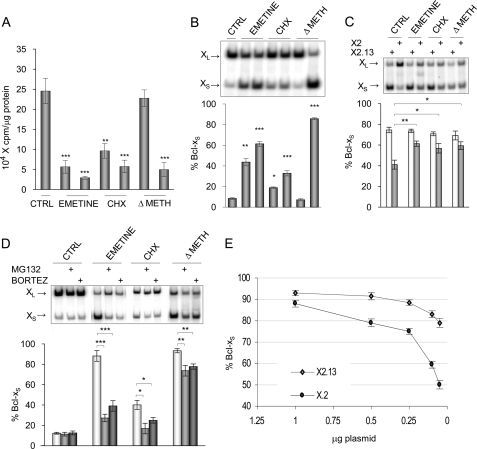
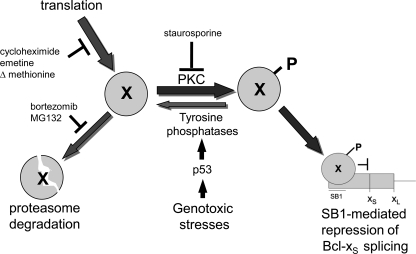
Alternative splicing often produces effectors with opposite functions in apoptosis. Splicing decisions must therefore be tightly connected to stresses, stimuli, and pathways that control cell survival and cell growth. We have shown previously that PKC signaling prevents the production of proapoptotic Bcl-x(S) to favor the accumulation of the larger antiapoptotic Bcl-x(L) splice variant in 293 cells. Here we show that the genotoxic stress induced by oxaliplatin elicits an ATM-, CHK2-, and p53-dependent splicing switch that favors the production of the proapoptotic Bcl-x(S) variant. This DNA damage-induced splicing shift requires the activity of protein-tyrosine phosphatases. Interestingly, the ATM/CHK2/p53/tyrosine phosphatases pathway activated by oxaliplatin regulates Bcl-x splicing through the same regulatory sequence element (SB1) that receives signals from the PKC pathway. Convergence of the PKC and DNA damage signaling routes may control the abundance of a key splicing repressor because SB1-mediated repression is lost when protein synthesis is impaired but is rescued by blocking proteasome-mediated protein degradation. The SB1 splicing regulatory module therefore receives antagonistic signals from the PKC and the p53-dependent DNA damage response pathways to control the balance of pro- and antiapoptotic Bcl-x splice variants.
Figures
Similar articles
-
Aberrant Bcl-x splicing in cancer: from molecular mechanism to therapeutic modulation.J Exp Clin Cancer Res. 2021 Jun 12;40(1):194. doi: 10.1186/s13046-021-02001-w. J Exp Clin Cancer Res. 2021. PMID: 34118966 Free PMC article. Review.
-
SRSF10 Connects DNA Damage to the Alternative Splicing of Transcripts Encoding Apoptosis, Cell-Cycle Control, and DNA Repair Factors.Cell Rep. 2016 Nov 15;17(8):1990-2003. doi: 10.1016/j.celrep.2016.10.071. Cell Rep. 2016. PMID: 27851963 Free PMC article.
-
Protein kinase C-dependent control of Bcl-x alternative splicing.Mol Cell Biol. 2007 Dec;27(24):8431-41. doi: 10.1128/MCB.00565-07. Epub 2007 Oct 8. Mol Cell Biol. 2007. PMID: 17923691 Free PMC article.
-
The Proto-oncogene PKCι regulates the alternative splicing of Bcl-x pre-mRNA.Mol Cancer Res. 2012 May;10(5):660-9. doi: 10.1158/1541-7786.MCR-11-0363. Epub 2012 Apr 20. Mol Cancer Res. 2012. PMID: 22522453 Free PMC article.
-
My road to alternative splicing control: from simple paths to loops and interconnections.Biochem Cell Biol. 2015 Jun;93(3):171-9. doi: 10.1139/bcb-2014-0161. Epub 2015 Feb 12. Biochem Cell Biol. 2015. PMID: 25759250 Review.
Cited by
-
Molecular prediction of adjuvant cisplatin efficacy in Non-Small Cell Lung Cancer (NSCLC)-validation in two independent cohorts.PLoS One. 2018 Mar 22;13(3):e0194609. doi: 10.1371/journal.pone.0194609. eCollection 2018. PLoS One. 2018. PMID: 29566065 Free PMC article.
-
Pre-mRNA processing factors meet the DNA damage response.Front Genet. 2013 Jun 6;4:102. doi: 10.3389/fgene.2013.00102. eCollection 2013. Front Genet. 2013. PMID: 23761808 Free PMC article.
-
Aberrant Bcl-x splicing in cancer: from molecular mechanism to therapeutic modulation.J Exp Clin Cancer Res. 2021 Jun 12;40(1):194. doi: 10.1186/s13046-021-02001-w. J Exp Clin Cancer Res. 2021. PMID: 34118966 Free PMC article. Review.
-
SRSF10 Connects DNA Damage to the Alternative Splicing of Transcripts Encoding Apoptosis, Cell-Cycle Control, and DNA Repair Factors.Cell Rep. 2016 Nov 15;17(8):1990-2003. doi: 10.1016/j.celrep.2016.10.071. Cell Rep. 2016. PMID: 27851963 Free PMC article.
-
The RNA Splicing Response to DNA Damage.Biomolecules. 2015 Oct 29;5(4):2935-77. doi: 10.3390/biom5042935. Biomolecules. 2015. PMID: 26529031 Free PMC article. Review.
References
-
- Adams J. M. (2003) Genes Dev. 17, 2481–2495 - PubMed
-
- Green D. R., Kroemer G. (2004) Science 305, 626–629 - PubMed
-
- Schwerk C., Schulze-Osthoff K. (2005) Mol. Cell 19, 1–13 - PubMed
-
- Jiang Z. H., Wu J. Y. (1999) Proc. Soc. Exp. Biol. Med. 220, 64–72 - PubMed
-
- Wu J. Y., Tang H., Havlioglu N. (2003) Prog. Mol. Subcell Biol. 31, 153–185 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
