

Giant virus with a remarkable complement of genes infects marine zooplankton
- PMID: 20974979
- PMCID: PMC2984142
- DOI: 10.1073/pnas.1007615107
Giant virus with a remarkable complement of genes infects marine zooplankton
Abstract
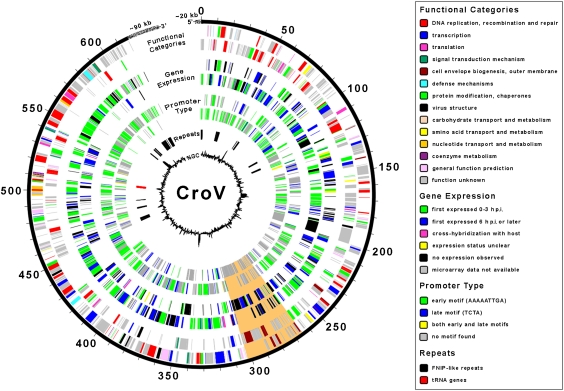
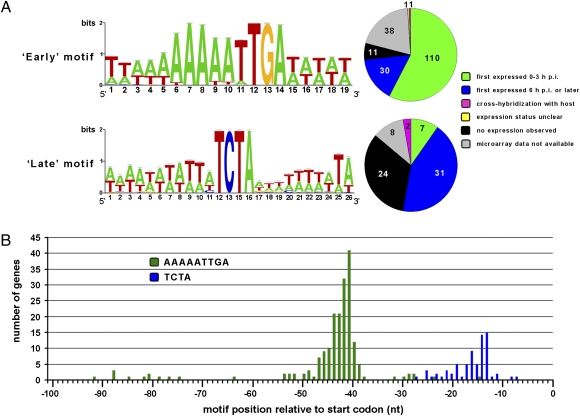
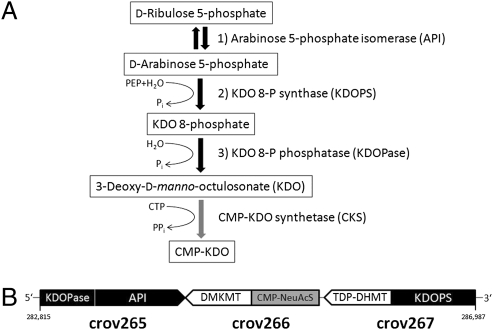
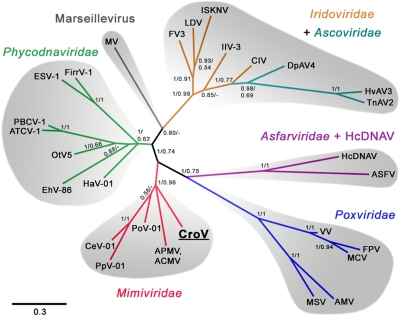
As major consumers of heterotrophic bacteria and phytoplankton, microzooplankton are a critical link in aquatic foodwebs. Here, we show that a major marine microflagellate grazer is infected by a giant virus, Cafeteria roenbergensis virus (CroV), which has the largest genome of any described marine virus (≈730 kb of double-stranded DNA). The central 618-kb coding part of this AT-rich genome contains 544 predicted protein-coding genes; putative early and late promoter motifs have been detected and assigned to 191 and 72 of them, respectively, and at least 274 genes were expressed during infection. The diverse coding potential of CroV includes predicted translation factors, DNA repair enzymes such as DNA mismatch repair protein MutS and two photolyases, multiple ubiquitin pathway components, four intein elements, and 22 tRNAs. Many genes including isoleucyl-tRNA synthetase, eIF-2γ, and an Elp3-like histone acetyltransferase are usually not found in viruses. We also discovered a 38-kb genomic region of putative bacterial origin, which encodes several predicted carbohydrate metabolizing enzymes, including an entire pathway for the biosynthesis of 3-deoxy-d-manno-octulosonate, a key component of the outer membrane in Gram-negative bacteria. Phylogenetic analysis indicates that CroV is a nucleocytoplasmic large DNA virus, with Acanthamoeba polyphaga mimivirus as its closest relative, although less than one-third of the genes of CroV have homologs in Mimivirus. CroV is a highly complex marine virus and the only virus studied in genetic detail that infects one of the major groups of predators in the oceans.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Another really, really big virus.Viruses. 2011 Jan;3(1):32-46. doi: 10.3390/v3010032. Epub 2011 Jan 18. Viruses. 2011. PMID: 21994725 Free PMC article.
-
The virion of Cafeteria roenbergensis virus (CroV) contains a complex suite of proteins for transcription and DNA repair.Virology. 2014 Oct;466-467:82-94. doi: 10.1016/j.virol.2014.05.029. Epub 2014 Jun 25. Virology. 2014. PMID: 24973308
-
Isolation and Identification of a Large Green Alga Virus (Chlorella Virus XW01) of Mimiviridae and Its Virophage (Chlorella Virus Virophage SW01) by Using Unicellular Green Algal Cultures.J Virol. 2022 Apr 13;96(7):e0211421. doi: 10.1128/jvi.02114-21. Epub 2022 Mar 9. J Virol. 2022. PMID: 35262372 Free PMC article.
-
Evolutionary genomics of nucleo-cytoplasmic large DNA viruses.Virus Res. 2006 Apr;117(1):156-84. doi: 10.1016/j.virusres.2006.01.009. Epub 2006 Feb 21. Virus Res. 2006. PMID: 16494962 Review.
-
Mimivirus and the emerging concept of "giant" virus.Virus Res. 2006 Apr;117(1):133-44. doi: 10.1016/j.virusres.2006.01.008. Epub 2006 Feb 15. Virus Res. 2006. PMID: 16469402 Review.
Cited by
-
Crystal structures of FNIP/FGxxFN motif-containing leucine-rich repeat proteins.Sci Rep. 2022 Sep 30;12(1):16430. doi: 10.1038/s41598-022-20758-8. Sci Rep. 2022. PMID: 36180492 Free PMC article.
-
Covariance of Marine Nucleocytoplasmic Large DNA Viruses with Eukaryotic Plankton Communities in the Sub-Arctic Kongsfjorden Ecosystem: A Metagenomic Analysis of Marine Microbial Ecosystems.Microorganisms. 2023 Jan 9;11(1):169. doi: 10.3390/microorganisms11010169. Microorganisms. 2023. PMID: 36677461 Free PMC article.
-
Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences.Open Biol. 2013 Dec 11;3(12):130160. doi: 10.1098/rsob.130160. Open Biol. 2013. PMID: 24335607 Free PMC article.
-
Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae.Proc Natl Acad Sci U S A. 2011 Oct 18;108(42):17486-91. doi: 10.1073/pnas.1110889108. Epub 2011 Oct 10. Proc Natl Acad Sci U S A. 2011. PMID: 21987820 Free PMC article.
-
Complete genome sequence of Courdo11 virus, a member of the family Mimiviridae.Virus Genes. 2014 Apr;48(2):218-23. doi: 10.1007/s11262-013-1016-x. Epub 2013 Dec 1. Virus Genes. 2014. PMID: 24293219
References
-
- Pernthaler J. Predation on prokaryotes in the water column and its ecological implications. Nat Rev Microbiol. 2005;3:537–546. - PubMed
-
- Raoult D, et al. The 1.2-megabase genome sequence of Mimivirus. Science. 2004;306:1344–1350. - PubMed
-
- Moreira D, López-García P. Ten reasons to exclude viruses from the tree of life. Nat Rev Microbiol. 2009;7:306–311. - PubMed
MeSH terms
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
