

Cellular metabolic stress: considering how cells respond to nutrient excess
- PMID: 20965425
- PMCID: PMC3190402
- DOI: 10.1016/j.molcel.2010.10.004
Cellular metabolic stress: considering how cells respond to nutrient excess
Abstract
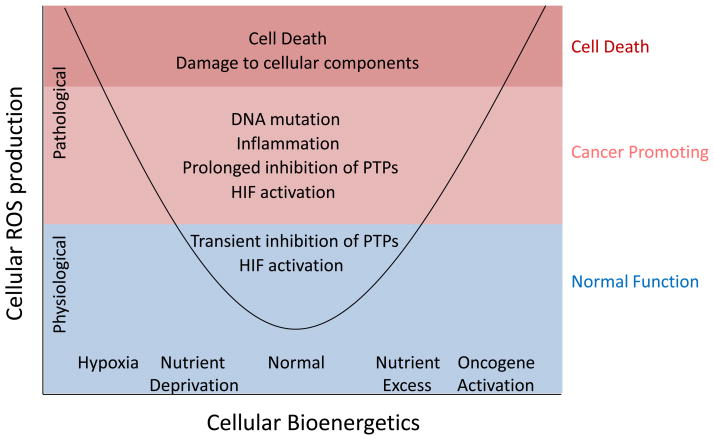
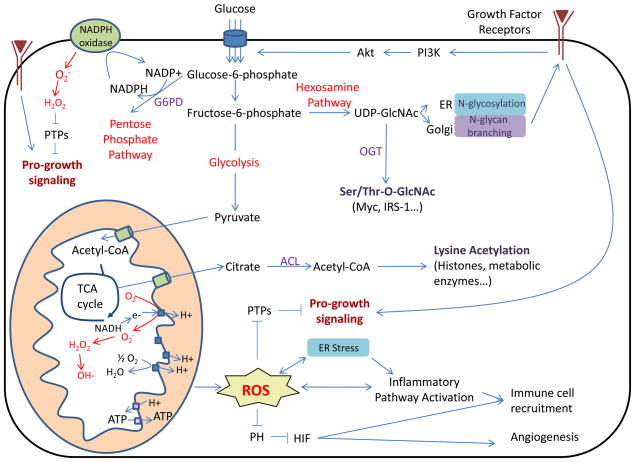
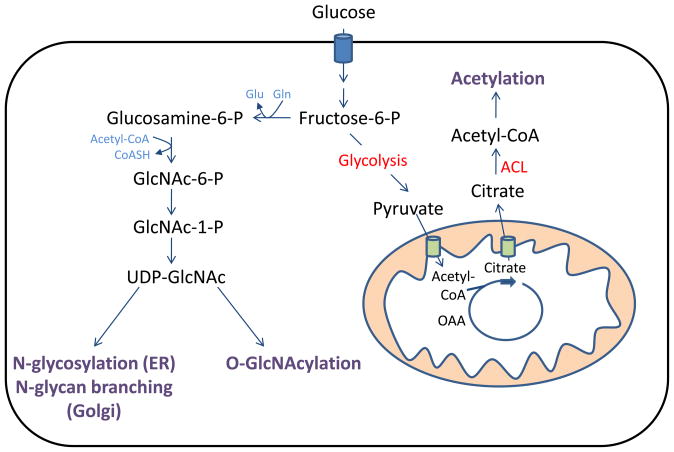
Nutrient stress is generally considered from the standpoint of how cells detect and respond to an insufficient supply of nutrients to meet their bioenergetic needs. However, cells also experience stress as a result of nutrient excess, during which reactive oxygen species (ROS) production exceeds that required for normal physiological responses. This may occur as a result of oncogene activation or chronic exposure to growth factors combined with high levels of nutrients. As a result, multiple mechanisms have evolved to allow cells to detect and adapt to elevated levels of intracellular metabolites, including promotion of signaling and proliferation by ROS, amino acid-dependent mTOR activation, and regulation of signaling and transcription through metabolite-sensitive protein modifications. We discuss how each of these responses can contribute to the development and/or progression of cancer under conditions of cellular nutrient excess and their potential roles in linking chronic organismal over-nutrition (obesity) with cancer.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway.Mol Cancer. 2017 Apr 13;16(1):79. doi: 10.1186/s12943-017-0648-1. Mol Cancer. 2017. PMID: 28407774 Free PMC article. Review.
-
Sirtuins and the Metabolic Hurdles in Cancer.Curr Biol. 2015 Jun 29;25(13):R569-83. doi: 10.1016/j.cub.2015.05.012. Curr Biol. 2015. PMID: 26126285 Free PMC article. Review.
-
[Adaptive response of cancer cells to metabolic stress].Nihon Rinsho. 2014 Jun;72(6):1058-62. Nihon Rinsho. 2014. PMID: 25016804 Japanese.
-
Maintaining homeostasis by controlled alternatives for energy distribution in plant cells under changing conditions of supply and demand.Photosynth Res. 2019 Mar;139(1-3):81-91. doi: 10.1007/s11120-018-0583-z. Epub 2018 Sep 10. Photosynth Res. 2019. PMID: 30203365 Free PMC article.
-
Is REDD1 a metabolic double agent? Lessons from physiology and pathology.Am J Physiol Cell Physiol. 2020 Nov 1;319(5):C807-C824. doi: 10.1152/ajpcell.00340.2020. Epub 2020 Sep 2. Am J Physiol Cell Physiol. 2020. PMID: 32877205 Review.
Cited by
-
Metabolic stress in autophagy and cell death pathways.Cold Spring Harb Perspect Biol. 2012 Sep 1;4(9):a008763. doi: 10.1101/cshperspect.a008763. Cold Spring Harb Perspect Biol. 2012. PMID: 22952396 Free PMC article. Review.
-
Identification of ubiquinol cytochrome c reductase hinge (UQCRH) as a potential diagnostic biomarker for lung adenocarcinoma.Open Biol. 2016 Jun;6(6):150256. doi: 10.1098/rsob.150256. Open Biol. 2016. PMID: 27358292 Free PMC article.
-
Tauroursodeoxycholic acid/TGR5 signaling promotes survival and early development of glucose-stressed porcine embryos†.Biol Reprod. 2021 Jul 2;105(1):76-86. doi: 10.1093/biolre/ioab072. Biol Reprod. 2021. PMID: 33889948 Free PMC article.
-
The Interlinking Metabolic Association between Type 2 Diabetes Mellitus and Cancer: Molecular Mechanisms and Therapeutic Insights.Diagnostics (Basel). 2024 Sep 25;14(19):2132. doi: 10.3390/diagnostics14192132. Diagnostics (Basel). 2024. PMID: 39410536 Free PMC article. Review.
-
ASB6 Promotes the Stemness Properties and Sustains Metastatic Potential of Oral Squamous Cell Carcinoma Cells by Attenuating ER Stress.Int J Biol Sci. 2019 Apr 22;15(5):1080-1090. doi: 10.7150/ijbs.31484. eCollection 2019. Int J Biol Sci. 2019. PMID: 31182927 Free PMC article.
References
-
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
