

Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl
- PMID: 20876800
- PMCID: PMC2955844
- DOI: 10.1158/0008-5472.CAN-10-0608
Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl
Abstract
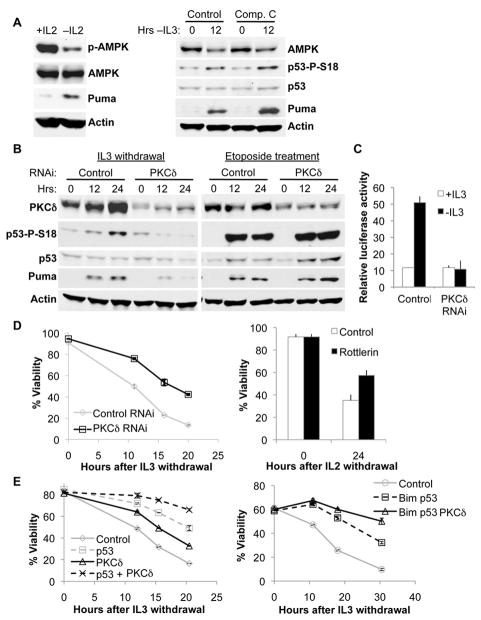
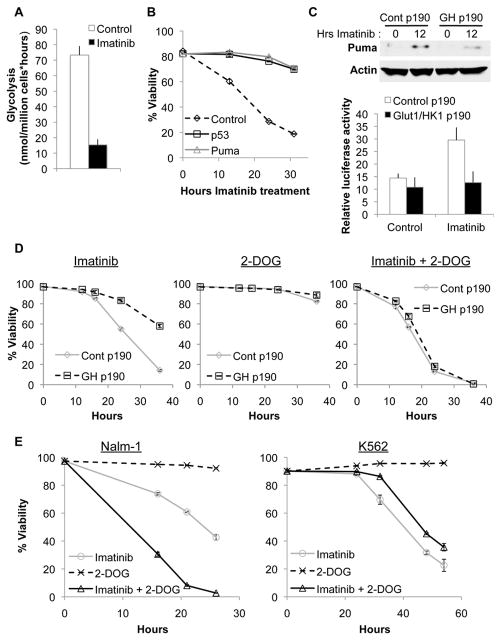
Unlike the growth factor dependence of normal cells, cancer cells can maintain growth factor-independent glycolysis and survival through expression of oncogenic kinases, such as BCR-Abl. Although targeted kinase inhibition can promote cancer cell death, therapeutic resistance develops frequently, and further mechanistic understanding is needed. Cell metabolism may be central to this cell death pathway, as we have shown that growth factor deprivation leads to decreased glycolysis that promotes apoptosis via p53 activation and induction of the proapoptotic protein Puma. Here, we extend these findings to show that elevated glucose metabolism, characteristic of cancer cells, can suppress protein kinase Cδ (PKCδ)-dependent p53 activation to maintain cell survival after growth factor withdrawal. In contrast, DNA damage-induced p53 activation was PKCδ independent and was not metabolically sensitive. Both stresses required p53 Ser(18) phosphorylation for maximal activity but led to unique patterns of p53 target gene expression, showing distinct activation and response pathways for p53 that were differentially regulated by metabolism. Consistent with oncogenic kinases acting to replace growth factors, treatment of BCR-Abl-expressing cells with the kinase inhibitor imatinib led to reduced metabolism and p53- and Puma-dependent cell death. Accordingly, maintenance of glucose uptake inhibited p53 activation and promoted imatinib resistance. Furthermore, inhibition of glycolysis enhanced imatinib sensitivity in BCR-Abl-expressing cells with wild-type p53 but had little effect on p53-null cells. These data show that distinct pathways regulate p53 after DNA damage and metabolic stress and that inhibiting glucose metabolism may enhance the efficacy of and overcome resistance to targeted molecular cancer therapies.
©2010 AACR.
Figures
Similar articles
-
Oncogenic stress induced by acute hyper-activation of Bcr-Abl leads to cell death upon induction of excessive aerobic glycolysis.PLoS One. 2011;6(9):e25139. doi: 10.1371/journal.pone.0025139. Epub 2011 Sep 20. PLoS One. 2011. PMID: 21949869 Free PMC article.
-
Imatinib mesylate induces cisplatin hypersensitivity in Bcr-Abl+ cells by differential modulation of p53 transcriptional and proapoptotic activity.Cancer Res. 2009 Dec 15;69(24):9337-45. doi: 10.1158/0008-5472.CAN-09-0548. Cancer Res. 2009. PMID: 19934315
-
Requirement for Mdm2 in the survival effects of Bcr-Abl and interleukin 3 in hematopoietic cells.Cancer Res. 2001 Oct 15;61(20):7635-41. Cancer Res. 2001. PMID: 11606405
-
C-Abl as a modulator of p53.Biochem Biophys Res Commun. 2005 Jun 10;331(3):737-49. doi: 10.1016/j.bbrc.2005.03.152. Biochem Biophys Res Commun. 2005. PMID: 15865930 Review.
-
A systematic review of p53 regulation of oxidative stress in skeletal muscle.Redox Rep. 2018 Dec;23(1):100-117. doi: 10.1080/13510002.2017.1416773. Epub 2018 Jan 3. Redox Rep. 2018. PMID: 29298131 Free PMC article. Review.
Cited by
-
Monocarboxylate transporter-1 promotes osteoblast differentiation via suppression of p53, a negative regulator of osteoblast differentiation.Sci Rep. 2018 Jul 12;8(1):10579. doi: 10.1038/s41598-018-28605-5. Sci Rep. 2018. PMID: 30002387 Free PMC article.
-
Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death.J Cell Biol. 2015 Aug 31;210(5):705-16. doi: 10.1083/jcb.201503044. J Cell Biol. 2015. PMID: 26323688 Free PMC article.
-
mRNA expression in papillary and anaplastic thyroid carcinoma: molecular anatomy of a killing switch.PLoS One. 2012;7(10):e37807. doi: 10.1371/journal.pone.0037807. Epub 2012 Oct 24. PLoS One. 2012. PMID: 23115614 Free PMC article.
-
Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition.Cancer Res. 2011 Aug 1;71(15):5204-13. doi: 10.1158/0008-5472.CAN-10-4531. Epub 2011 Jun 13. Cancer Res. 2011. PMID: 21670080 Free PMC article.
-
An R132H mutation in isocitrate dehydrogenase 1 enhances p21 expression and inhibits phosphorylation of retinoblastoma protein in glioma cells.Neurol Med Chir (Tokyo). 2013;53(10):645-54. doi: 10.2176/nmc.oa2012-0409. Epub 2013 Sep 27. Neurol Med Chir (Tokyo). 2013. PMID: 24077277 Free PMC article.
References
-
- Raff MC. Social controls on cell survival and cell death. Nature. 1992;356(6368):397–400. - PubMed
-
- Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6(3):683–92. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. - PubMed
-
- Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
