

Three phases of DiGeorge/22q11 deletion syndrome pathogenesis during brain development: patterning, proliferation, and mitochondrial functions of 22q11 genes
- PMID: 20833244
- PMCID: PMC3770287
- DOI: 10.1016/j.ijdevneu.2010.08.005
Three phases of DiGeorge/22q11 deletion syndrome pathogenesis during brain development: patterning, proliferation, and mitochondrial functions of 22q11 genes
Abstract
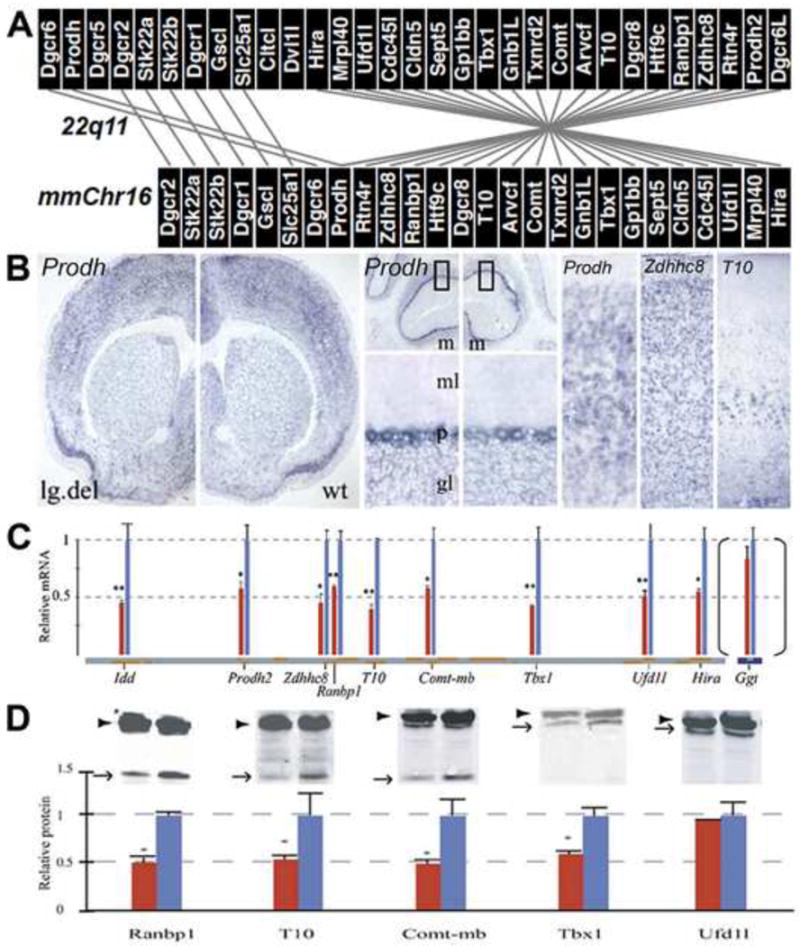
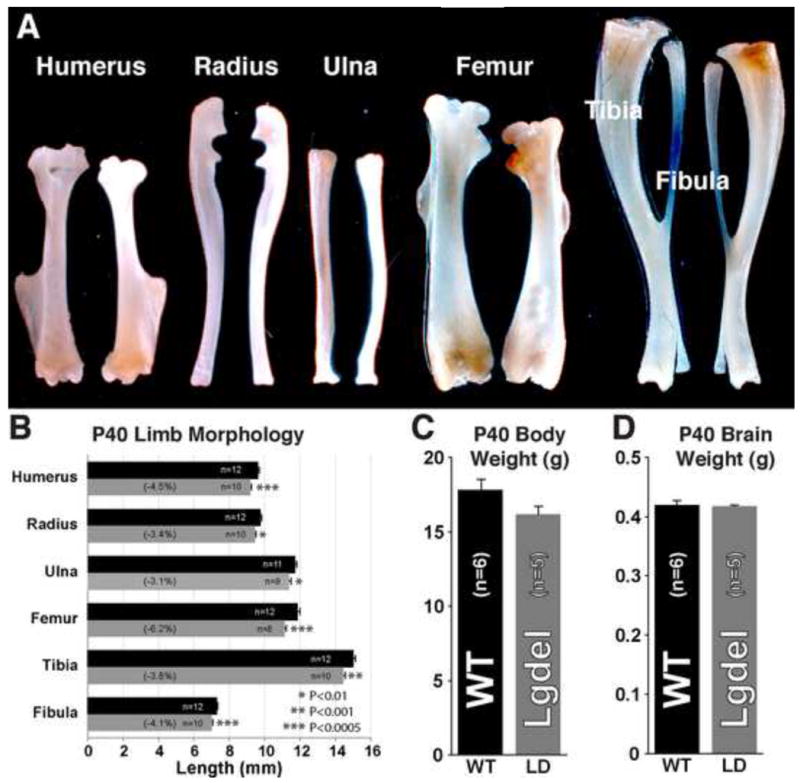
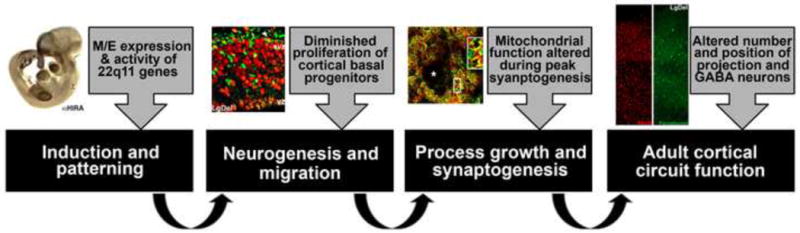
DiGeorge, or 22q11 deletion syndrome (22q11DS), the most common survivable human genetic deletion disorder, is caused by deletion of a minimum of 32 contiguous genes on human chromosome 22, and presumably results from diminished dosage of one, some, or all of these genes--particularly during development. Nevertheless, the normal functions of 22q11 genes in the embryo or neonate, and their contribution to developmental pathogenesis that must underlie 22q11DS are not well understood. Our data suggests that a substantial number of 22q11 genes act specifically and in concert to mediate early morphogenetic interactions and subsequent cellular differentiation at phenotypically compromised sites--the limbs, heart, face and forebrain. When dosage of a broad set of these genes is diminished, early morphogenesis is altered, and initial 22q11DS phenotypes are established. Thereafter, functionally similar subsets of 22q11 genes--especially those that influence the cell cycle or mitochondrial function--remain expressed, particularly in the developing cerebral cortex, to regulate neurogenesis and synaptic development. When dosage of these genes is diminished, numbers, placement and connectivity of neurons and circuits essential for normal behavior may be disrupted. Such disruptions likely contribute to vulnerability for schizophrenia, autism, or attention deficit/hyperactivity disorder seen in most 22q11DS patients.
Copyright © 2010 ISDN. Published by Elsevier Ltd. All rights reserved.
Figures





Similar articles
-
Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome.Proc Natl Acad Sci U S A. 2009 Sep 22;106(38):16434-45. doi: 10.1073/pnas.0905696106. Epub 2009 Sep 10. Proc Natl Acad Sci U S A. 2009. PMID: 19805316 Free PMC article.
-
In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: are they all suspects?J Neurodev Disord. 2019 Jun 7;11(1):7. doi: 10.1186/s11689-019-9267-z. J Neurodev Disord. 2019. PMID: 31174463 Free PMC article. Review.
-
Views of adults with 22q11 deletion syndrome on reproductive choices.Am J Med Genet A. 2020 May;182(5):1284-1287. doi: 10.1002/ajmg.a.61546. Epub 2020 Mar 10. Am J Med Genet A. 2020. PMID: 32154643 No abstract available.
-
Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes.Mol Cell Neurosci. 2008 Nov;39(3):439-51. doi: 10.1016/j.mcn.2008.07.027. Epub 2008 Aug 15. Mol Cell Neurosci. 2008. PMID: 18775783 Free PMC article.
-
When half is not enough: gene expression and dosage in the 22q11 deletion syndrome.Gene Expr. 2007;13(6):299-310. doi: 10.3727/000000006781510697. Gene Expr. 2007. PMID: 17708416 Free PMC article. Review.
Cited by
-
Losing your inhibition: linking cortical GABAergic interneurons to schizophrenia.Neurobiol Dis. 2013 May;53:36-48. doi: 10.1016/j.nbd.2012.11.013. Epub 2012 Nov 29. Neurobiol Dis. 2013. PMID: 23201207 Free PMC article. Review.
-
The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis.Oncotarget. 2012 Oct;3(10):1220-35. doi: 10.18632/oncotarget.714. Oncotarget. 2012. PMID: 23100451 Free PMC article.
-
Screening of 22q11.2DS Using Multiplex Ligation-Dependent Probe Amplification as an Alternative Diagnostic Method.Biomed Res Int. 2020 Sep 28;2020:6945730. doi: 10.1155/2020/6945730. eCollection 2020. Biomed Res Int. 2020. PMID: 33062692 Free PMC article.
-
Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS.Cereb Cortex. 2015 May;25(5):1143-51. doi: 10.1093/cercor/bht308. Epub 2013 Nov 11. Cereb Cortex. 2015. PMID: 24217989 Free PMC article.
-
Mitochondrial Citrate Transporter-dependent Metabolic Signature in the 22q11.2 Deletion Syndrome.J Biol Chem. 2015 Sep 18;290(38):23240-53. doi: 10.1074/jbc.M115.672360. Epub 2015 Jul 28. J Biol Chem. 2015. PMID: 26221035 Free PMC article.
References
-
- Arnsten AF. Fundamentals of attention-deficit/hyperactivity disorder: circuits and pathways. J Clin Psychiatry. 2006;67(Suppl 8):7–12. - PubMed
-
- Bachiller D, Klingensmith J, Shneyder N, Tran U, Anderson R, Rossant J, De Robertis EM. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development. 2003;130:3567–3578. - PubMed
-
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–715. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
