

Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I
- PMID: 20810878
- PMCID: PMC2943838
- DOI: 10.1523/JNEUROSCI.1746-10.2010
Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I
Abstract
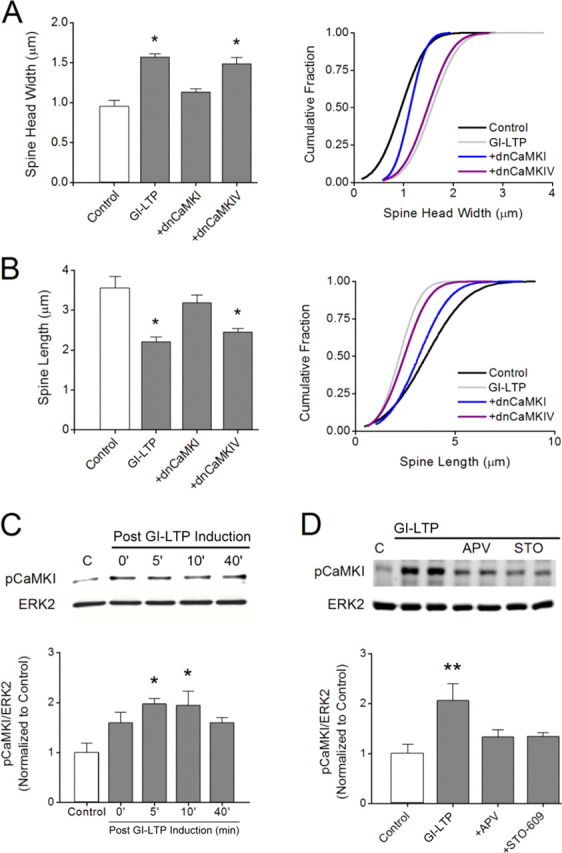
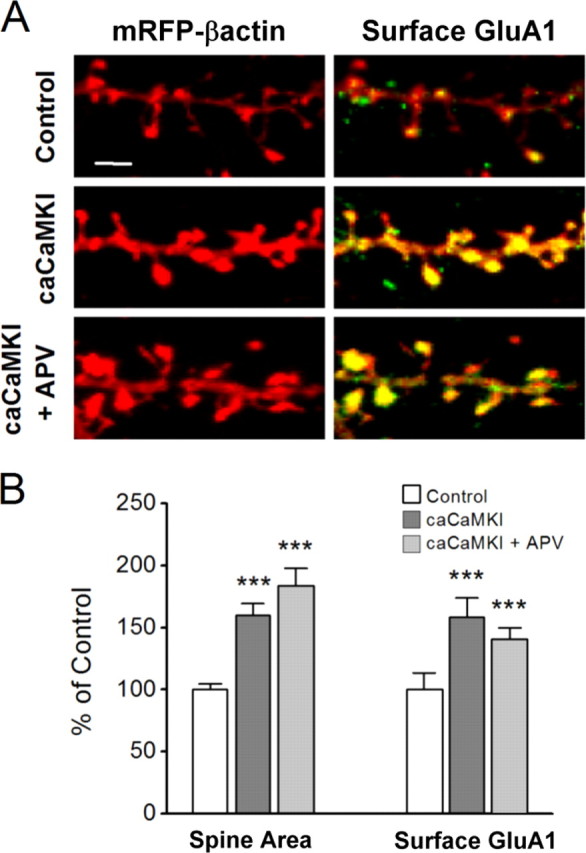
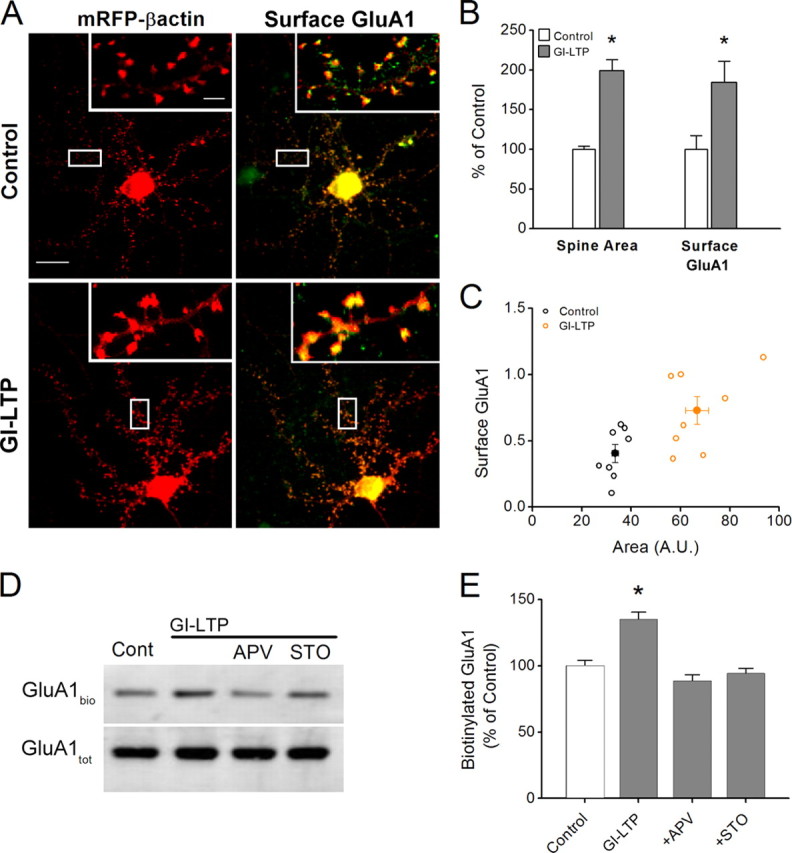
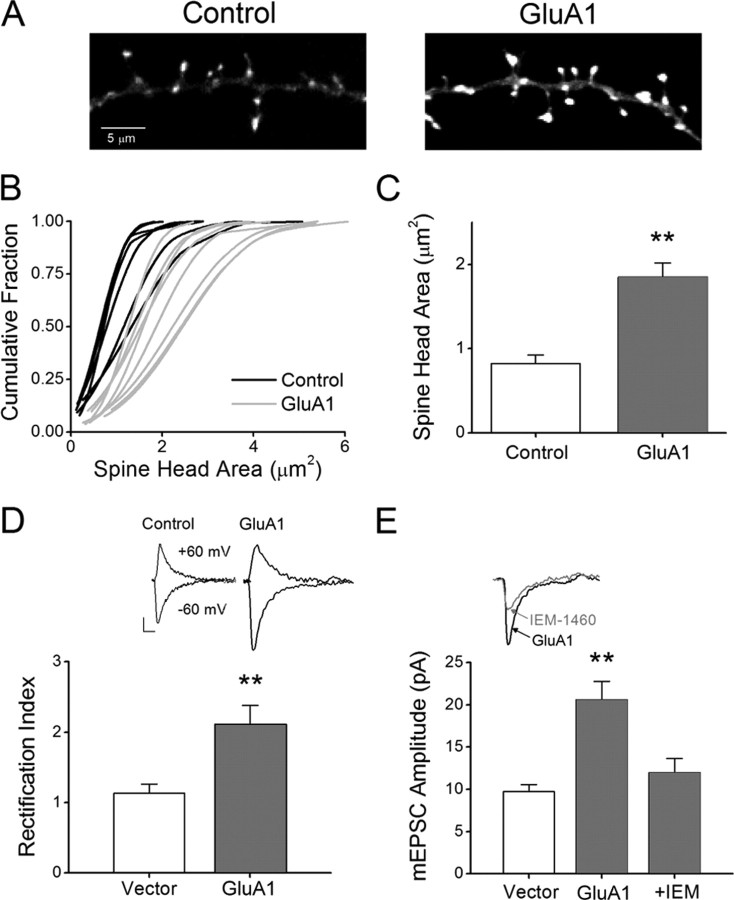
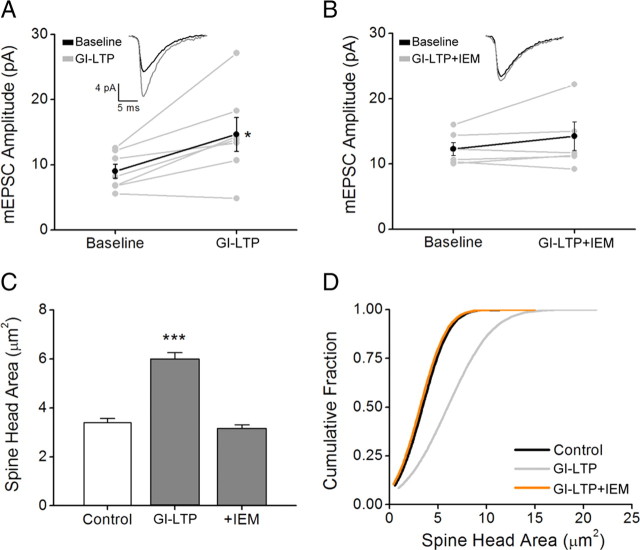
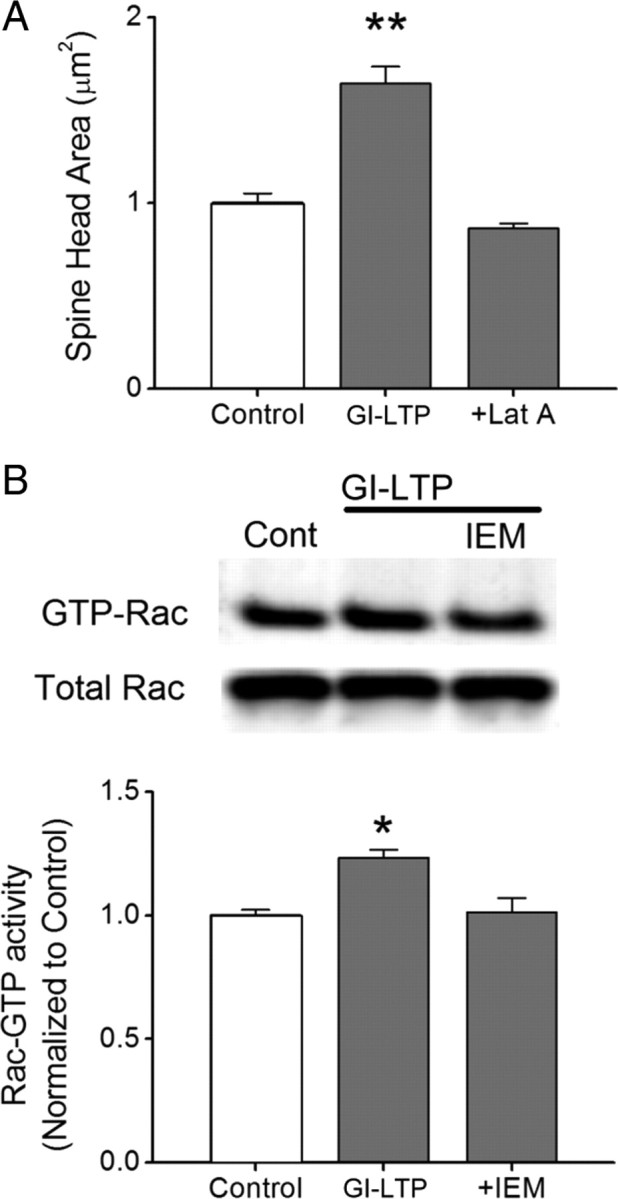
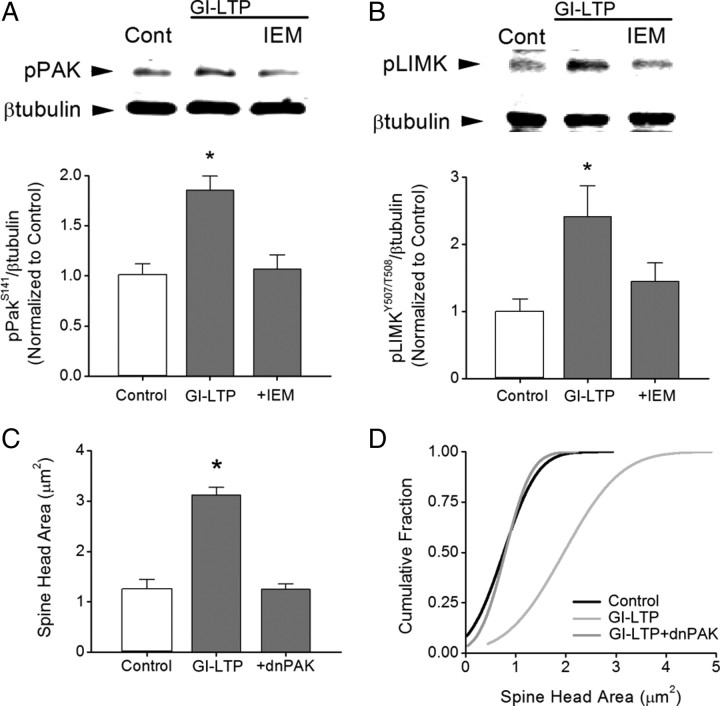
It is well established that long-term potentiation (LTP), a paradigm for learning and memory, results in a stable enlargement of potentiated spines associated with recruitment of additional GluA1-containing AMPA receptors (AMPARs). Although regulation of the actin cytoskeleton is involved, the detailed signaling mechanisms responsible for this spine expansion are unclear. Here, we used cultured mature hippocampal neurons stimulated with a glycine-induced, synapse-specific form of chemical LTP (GI-LTP). We report that the stable structural plasticity (i.e., spine head enlargement and spine length shortening) that accompanies GI-LTP was blocked by inhibitors of NMDA receptors (NMDARs; APV) or CaM-kinase kinase (STO-609), the upstream activator of CaM-kinase I (CaMKI), as well as by transfection with dominant-negative (dn) CaMKI but not dnCaMKIV. Recruitment of GluA1 to the spine surface occurred after GI-LTP and was mimicked by transfection with constitutively active CaMKI. Spine enlargement induced by transfection of GluA1 was associated with synaptic recruitment of Ca(2+)-permeable AMPARs (CP-AMPARs) as assessed by an increase in the rectification index of miniature EPSCs (mEPSCs) and their sensitivity to IEM-1460, a selective antagonist of CP-AMPARs. Furthermore, the increase in spine size and mEPSC amplitude resulting from GI-LTP itself was blocked by IEM-1460, demonstrating involvement of CP-AMPARs. Downstream signaling effectors of CP-AMPARs, identified by suppression of their activation by IEM-1460, included the Rac/PAK/LIM-kinase pathway that regulates spine actin dynamics. Together, our results suggest that synaptic recruitment of CP-AMPARs via CaMKI may provide a mechanistic link between NMDAR activation in LTP and regulation of a signaling pathway that drives spine enlargement via actin polymerization.
Figures

Similar articles
-
Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I.J Neurosci. 2008 Jun 4;28(23):6000-9. doi: 10.1523/JNEUROSCI.0384-08.2008. J Neurosci. 2008. PMID: 18524905 Free PMC article.
-
Control of Homeostatic Synaptic Plasticity by AKAP-Anchored Kinase and Phosphatase Regulation of Ca2+-Permeable AMPA Receptors.J Neurosci. 2018 Mar 14;38(11):2863-2876. doi: 10.1523/JNEUROSCI.2362-17.2018. Epub 2018 Feb 13. J Neurosci. 2018. PMID: 29440558 Free PMC article.
-
Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II.PLoS One. 2009;4(2):e4339. doi: 10.1371/journal.pone.0004339. Epub 2009 Feb 3. PLoS One. 2009. PMID: 19190753 Free PMC article.
-
Regulation of neuronal PKA signaling through AKAP targeting dynamics.Eur J Cell Biol. 2006 Jul;85(7):627-33. doi: 10.1016/j.ejcb.2006.01.010. Epub 2006 Feb 28. Eur J Cell Biol. 2006. PMID: 16504338 Review.
-
Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors.Crit Rev Neurobiol. 2006;18(1-2):71-84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. Crit Rev Neurobiol. 2006. PMID: 17725510 Review.
Cited by
-
The E. coli CNF1 as a pioneering therapy for the central nervous system diseases.Toxins (Basel). 2014 Jan 7;6(1):270-82. doi: 10.3390/toxins6010270. Toxins (Basel). 2014. PMID: 24402235 Free PMC article. Review.
-
Activity-dependent dendritic spine shrinkage and growth involve downregulation of cofilin via distinct mechanisms.PLoS One. 2014 Apr 16;9(4):e94787. doi: 10.1371/journal.pone.0094787. eCollection 2014. PLoS One. 2014. PMID: 24740405 Free PMC article.
-
Neuroprotection by ADAM10 inhibition requires TrkB signaling in the Huntington's disease hippocampus.Cell Mol Life Sci. 2024 Aug 7;81(1):333. doi: 10.1007/s00018-024-05382-1. Cell Mol Life Sci. 2024. PMID: 39112663 Free PMC article.
-
Molecular Mechanisms Underlying Ca2+/Calmodulin-Dependent Protein Kinase Kinase Signal Transduction.Int J Mol Sci. 2022 Sep 20;23(19):11025. doi: 10.3390/ijms231911025. Int J Mol Sci. 2022. PMID: 36232320 Free PMC article. Review.
-
Targeting of NF-κB to Dendritic Spines Is Required for Synaptic Signaling and Spine Development.J Neurosci. 2018 Apr 25;38(17):4093-4103. doi: 10.1523/JNEUROSCI.2663-16.2018. Epub 2018 Mar 19. J Neurosci. 2018. PMID: 29555853 Free PMC article.
References
-
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42.
-
- Ageta-Ishihara N, Takemoto-Kimura S, Nonaka M, Adachi-Morishima A, Suzuki K, Kamijo S, Fujii H, Mano T, Blaeser F, Chatila TA, Mizuno H, Hirano T, Tagawa Y, Okuno H, Bito H. Control of cortical axon elongation by a GABA-driven Ca2+/calmodulin-dependent protein kinase cascade. J Neurosci. 2009;29:13720–13729. - PMC - PubMed
-
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998;20:25–30. - PubMed
-
- Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999;9:364–370. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous