

Serum or target deprivation-induced neuronal death causes oxidative neuronal accumulation of Zn2+ and loss of NAD+
- PMID: 20722716
- PMCID: PMC2946389
- DOI: 10.1111/j.1460-9568.2010.07372.x
Serum or target deprivation-induced neuronal death causes oxidative neuronal accumulation of Zn2+ and loss of NAD+
Abstract
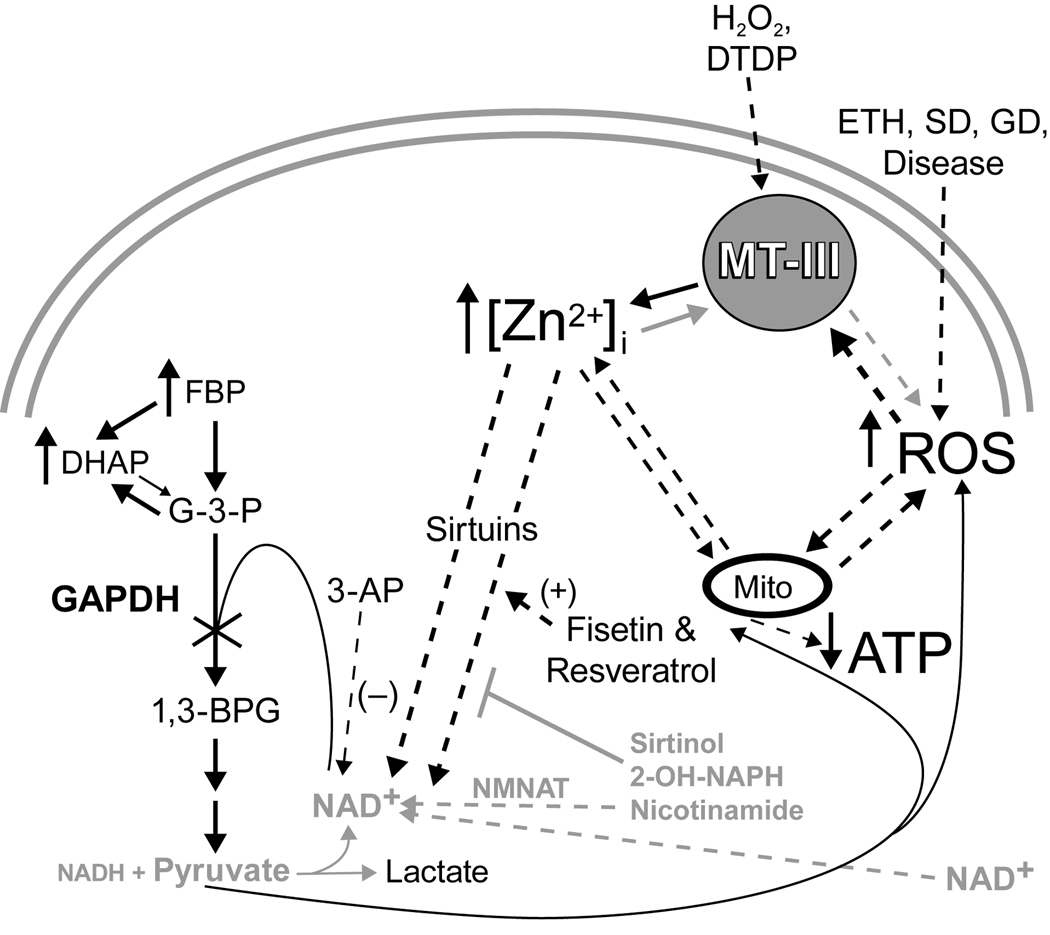
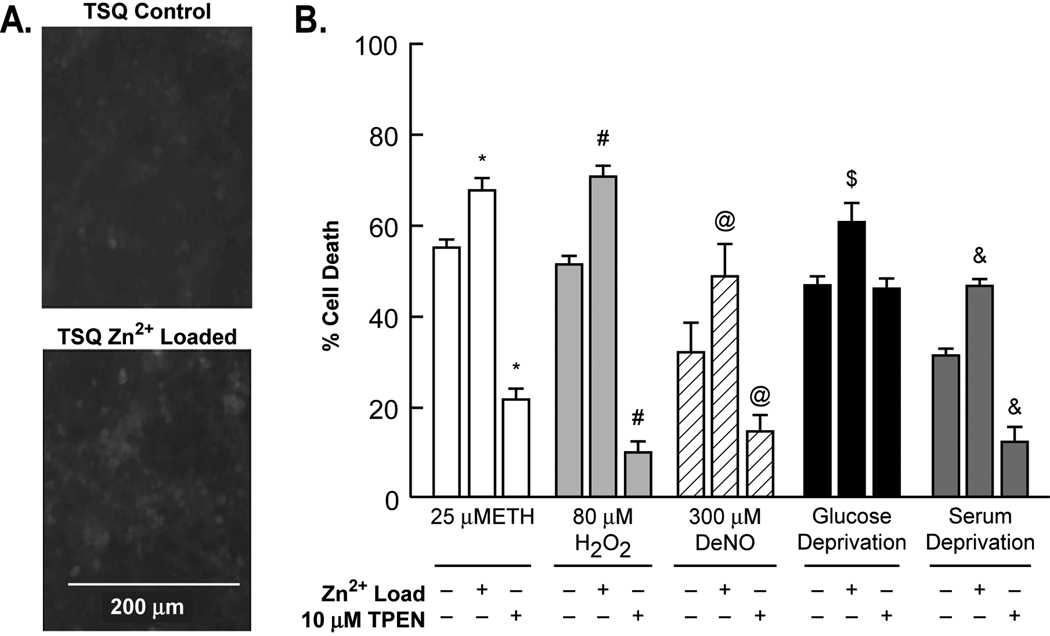
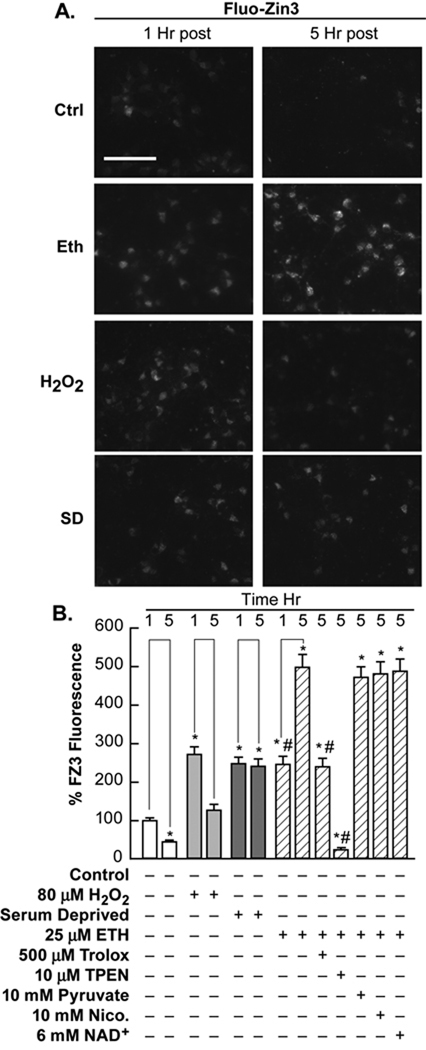
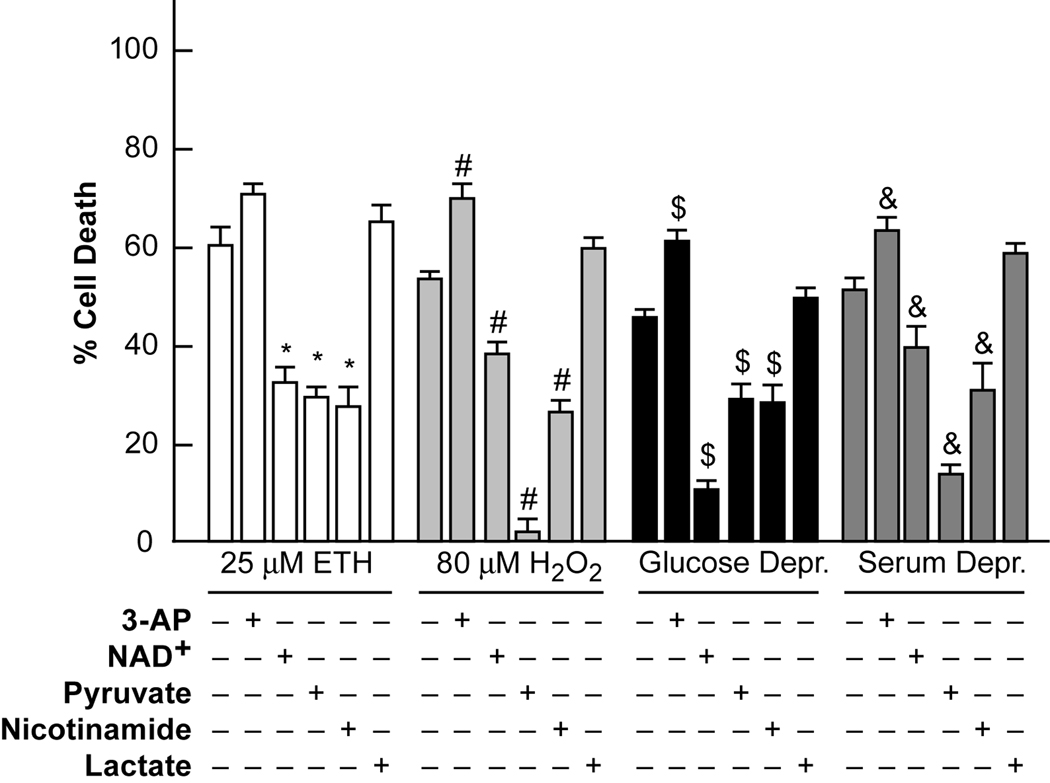
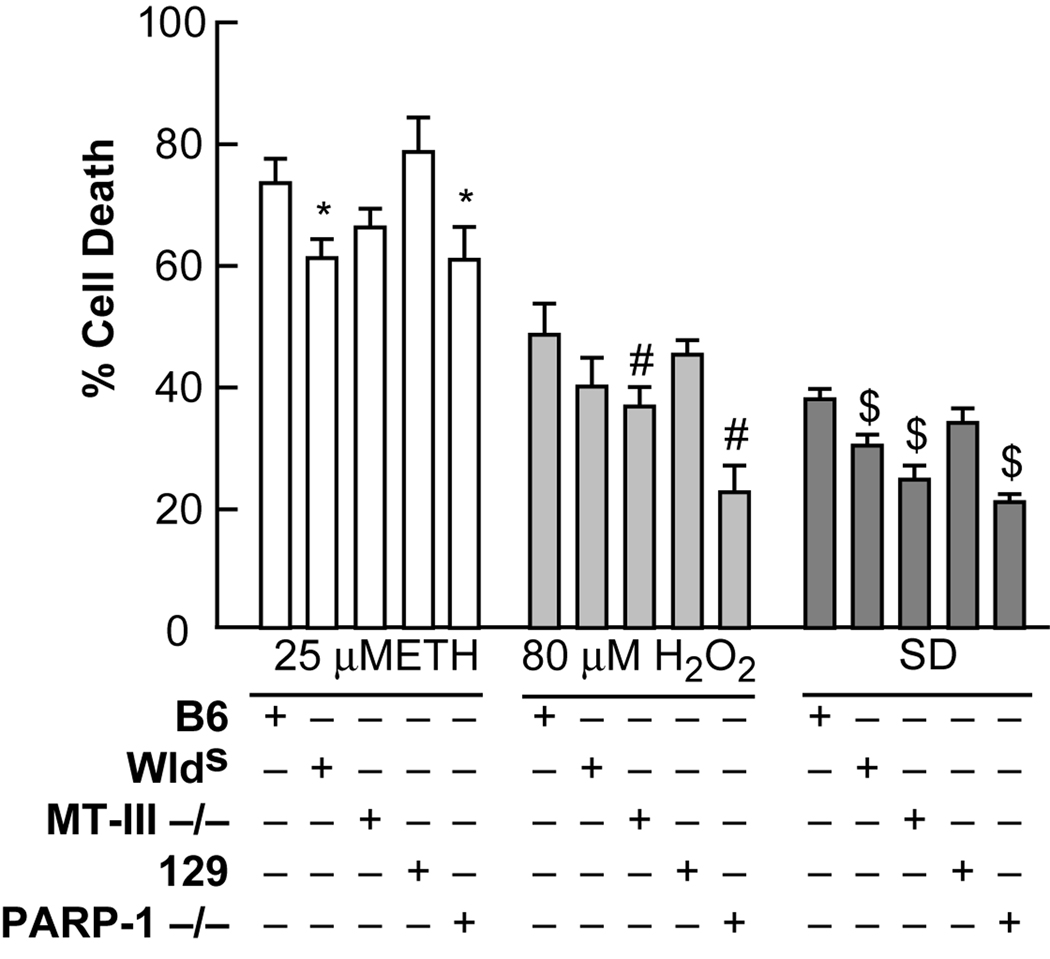
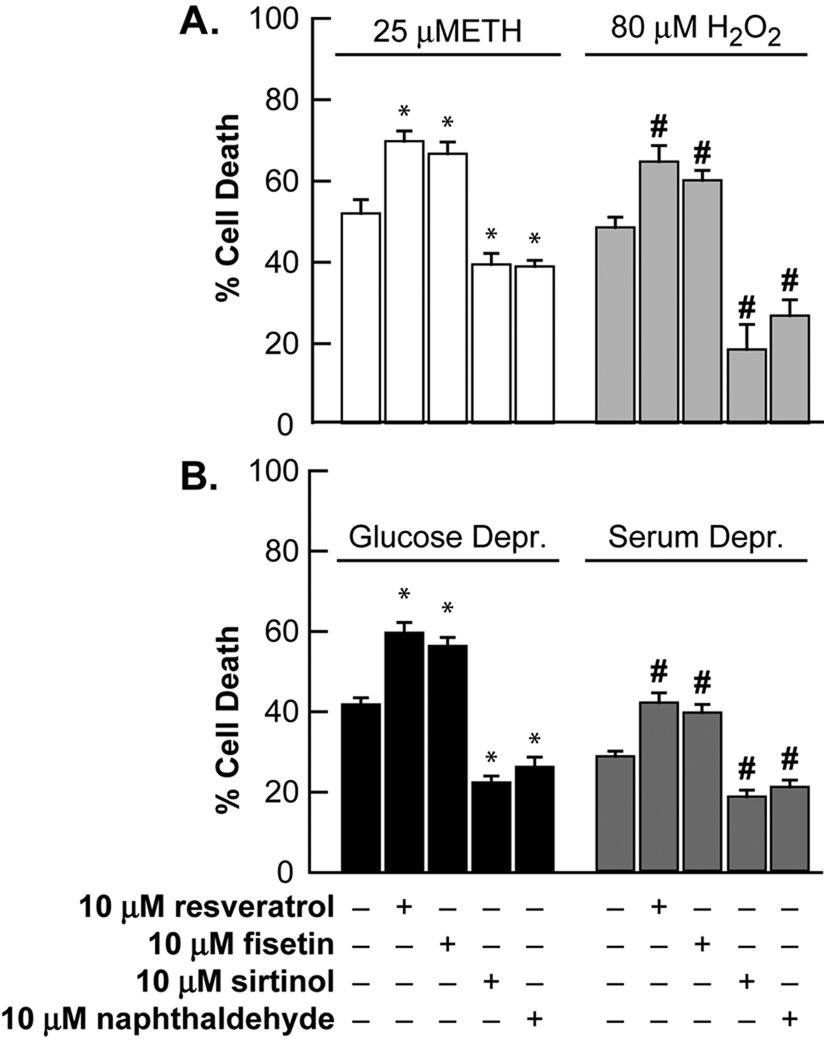
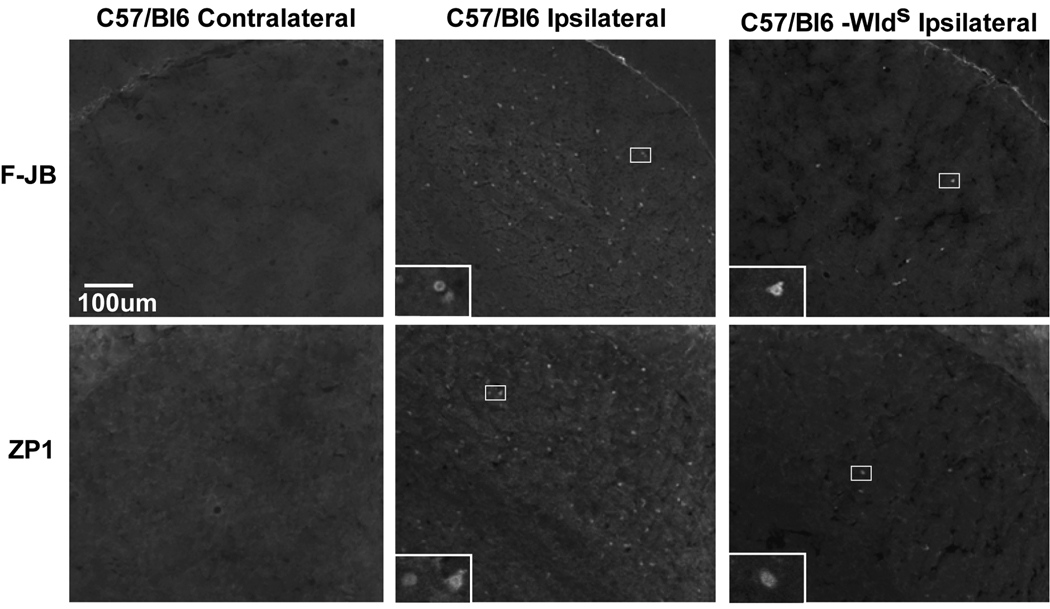
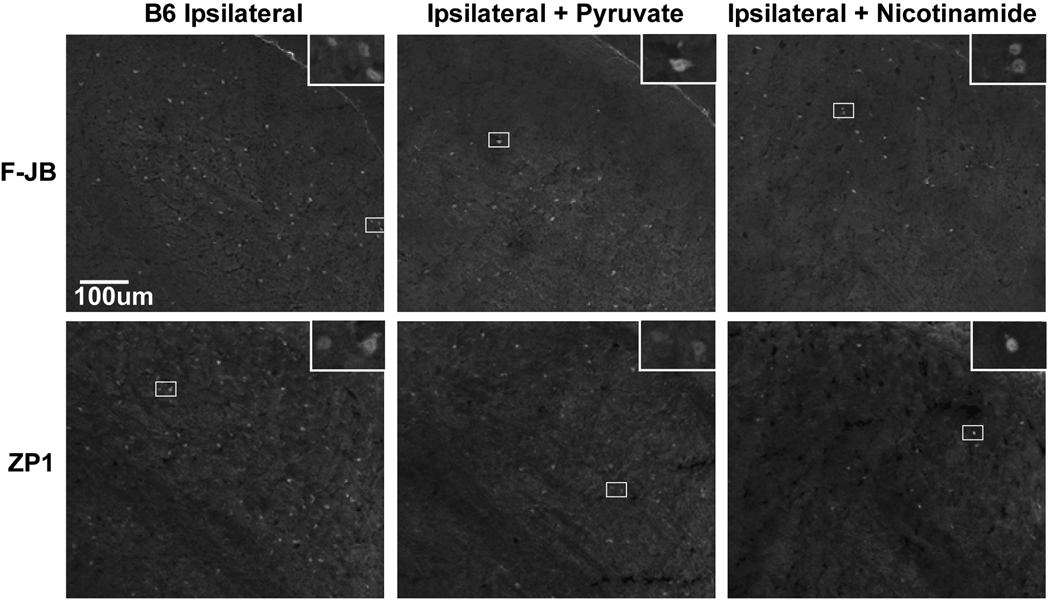
Trophic deprivation-mediated neuronal death is important during development, after acute brain or nerve trauma, and in neurodegeneration. Serum deprivation (SD) approximates trophic deprivation in vitro, and an in vivo model is provided by neuronal death in the mouse dorsal lateral geniculate nucleus (LGNd) after ablation of the visual cortex (VCA). Oxidant-induced intracellular Zn(2+) release ([Zn(2+) ](i) ) from metallothionein-3 (MT-III), mitochondria or 'protein Zn(2+) ', was implicated in trophic deprivation neurotoxicity. We have previously shown that neurotoxicity of extracellular Zn(2+) required entry, increased [Zn(2+) ](i) , and reduction of NAD(+) and ATP levels causing inhibition of glycolysis and cellular metabolism. Exogenous NAD(+) and sirtuin inhibition attenuated Zn(2+) neurotoxicity. Here we show that: (1) Zn(2+) is released intracellularly after oxidant and SD injuries, and that sensitivity to these injuries is proportional to neuronal Zn(2+) content; (2) NAD(+) loss is involved - restoration of NAD(+) using exogenous NAD(+) , pyruvate or nicotinamide attenuated these injuries, and potentiation of NAD(+) loss potentiated injury; (3) neurons from genetically modified mouse strains which reduce intracellular Zn(2+) content (MT-III knockout), reduce NAD(+) catabolism (PARP-1 knockout) or increase expression of an NAD(+) synthetic enzyme (Wld(s) ) each had attenuated SD and oxidant neurotoxicities; (4) sirtuin inhibitors attenuated and sirtuin activators potentiated these neurotoxicities; (5) visual cortex ablation (VCA) induces Zn(2+) staining and death only in ipsilateral LGNd neurons, and a 1 mg/kg Zn(2+) diet attenuated injury; and finally (6) NAD(+) synthesis and levels are involved given that LGNd neuronal death after VCA was dramatically reduced in Wld(s) animals, and by intraperitoneal pyruvate or nicotinamide. Zn(2+) toxicity is involved in serum and trophic deprivation-induced neuronal death.
© 2010 The Authors. European Journal of Neuroscience © 2010 Federation of European Neuroscience Societies and Blackwell Publishing Ltd.
Figures
Similar articles
-
Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway.Eur J Neurosci. 2006 Oct;24(8):2169-76. doi: 10.1111/j.1460-9568.2006.05110.x. Epub 2006 Oct 17. Eur J Neurosci. 2006. PMID: 17042794
-
Mitochondrial inhibitor models of Huntington's disease and Parkinson's disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo.Neurodegener Dis. 2013;11(1):49-58. doi: 10.1159/000336558. Epub 2012 May 24. Neurodegener Dis. 2013. PMID: 22627004
-
Light-induced photoreceptor and RPE degeneration involve zinc toxicity and are attenuated by pyruvate, nicotinamide, or cyclic light.Mol Vis. 2010 Dec 8;16:2639-52. Mol Vis. 2010. PMID: 21179242 Free PMC article.
-
NAD+ precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms.Exp Neurol. 2020 Mar;325:113144. doi: 10.1016/j.expneurol.2019.113144. Epub 2019 Dec 16. Exp Neurol. 2020. PMID: 31837320 Free PMC article. Review.
-
Redox regulation of intracellular zinc: molecular signaling in the life and death of neurons.Antioxid Redox Signal. 2011 Oct 15;15(8):2249-63. doi: 10.1089/ars.2010.3607. Epub 2011 Mar 31. Antioxid Redox Signal. 2011. PMID: 20849376 Free PMC article. Review.
Cited by
-
NAD+, Senolytics, or Pyruvate for Healthy Aging?Nutr Metab Insights. 2021 Oct 26;14:11786388211053407. doi: 10.1177/11786388211053407. eCollection 2021. Nutr Metab Insights. 2021. PMID: 34720589 Free PMC article. Review.
-
Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo.PLoS One. 2012;7(7):e42129. doi: 10.1371/journal.pone.0042129. Epub 2012 Jul 25. PLoS One. 2012. PMID: 22848730 Free PMC article.
-
Ion channels and zinc: mechanisms of neurotoxicity and neurodegeneration.J Toxicol. 2012;2012:785647. doi: 10.1155/2012/785647. Epub 2012 May 7. J Toxicol. 2012. PMID: 22645609 Free PMC article.
-
Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment.Biomolecules. 2020 Apr 29;10(5):687. doi: 10.3390/biom10050687. Biomolecules. 2020. PMID: 32365524 Free PMC article. Review.
-
Real-time impedance-based cell analyzer as a tool to delineate molecular pathways involved in neurotoxicity and neuroprotection in a neuronal cell line.J Vis Exp. 2014 Aug 9;(90):e51748. doi: 10.3791/51748. J Vis Exp. 2014. PMID: 25146163 Free PMC article.
References
-
- Agarwala S, Kalil RE. Axotomy-induced neuronal death and reactive astrogliosis in the lateral geniculate nucleus following a lesion of the visual cortex in the rat. J Comp Neurol. 1998;392:252–263. - PubMed
-
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. - PubMed
-
- Al-Abdulla NA, Portera-Cailliau C, Martin LJ. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience. 1998;86:191–209. - PubMed
-
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. - PubMed
-
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK073446/DK/NIDDK NIH HHS/United States
- R01 DK073446-05/DK/NIDDK NIH HHS/United States
- R01 DK073446-02/DK/NIDDK NIH HHS/United States
- R01 DK073446-06/DK/NIDDK NIH HHS/United States
- R01 NS030337-13/NS/NINDS NIH HHS/United States
- R01 DK073446-03/DK/NIDDK NIH HHS/United States
- R01 DK073446-05S2/DK/NIDDK NIH HHS/United States
- NS 030337/NS/NINDS NIH HHS/United States
- DK 073446/DK/NIDDK NIH HHS/United States
- R01 DK073446-04/DK/NIDDK NIH HHS/United States
- R01 DK073446-01A1/DK/NIDDK NIH HHS/United States
- R01 NS030337/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
